Plasma levels of sublingual buprenorphine utilized in the therapy of opioid use disorder, has been demonstrated to undergo gestation-associated decline in vivo, to an extent influenced by upheavals physiologically across gestational trimesters. However, based on extant literature, a dearth of knowledge exists in the optimization of buprenorphine therapeutic modalities, pharmacokinetic interactions and posological scrutiny, necessary for successful regimen adherence. A physiologically-based pharmacokinetic modelling methodology in a virtual clinical trial premise was utilized to investigate gestational upheavals in peak plasma buprenorphine concentrations, followed by a pharmacokinetic drug-drug interaction investigation and dose optimization strategy, to maintain buprenorphine levels above proposed thresholds of 1ng/ml and below 22.2ng/ml adjudicated as a fatality limit. A fold decline (> 1.3fold) in buprenorphine mean peak plasma concentration (92% - 74%) was evident for the model predicted buprenorphine metrics across selected gestational weeks to term in line with the model predicted increases in physiological upheavals occurring across gestation which may influence the changes. The rifampicin mediated drug-drug interaction on buprenorphine levels initially resulted in fold decreases (>1.5 fold) over a twenty-four hour duration, in concert with escalating physiological metrics across gestational trimesters. The interaction perpetrated with Clarithromycin dosing resulted in fold increases (> 2-fold) in the plasma concentration as well as an increase in other metrics associated with buprenorphine kinetics. The dose optimization approach maintained majority of subjects (>90%) with the extensive metabolizer (EM) phenotype above 1ng/ml and below 22.2ng/ml in the 8mg – 24mg dose ranges albeit with 1% and 3% in the 28mg and 32mg doses above the fatality limit respectively. This study demonstrates the utility of physiologically based pharmacokinetic methods to predict the time course of administered buprenorphine in plasma during gestation which could aid clinician decisions in a translational manner, in order to optimize therapeutic modalities in the therapy of opioid use disorder.
| Published in | International Journal of Pharmacy and Chemistry (Volume 10, Issue 4) |
| DOI | 10.11648/j.ijpc.20241004.11 |
| Page(s) | 46-79 |
| Creative Commons |
This is an Open Access article, distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution and reproduction in any medium or format, provided the original work is properly cited. |
| Copyright |
Copyright © The Author(s), 2024. Published by Science Publishing Group |
Buprenorphine, Drug Interactions, Dose, Optimization, Pharmacokinetics, Phenotype, Pregnancy, PBPK
N | Mean age (range) | Dose (mg) | AUC | Cmax | Tmax (h) | |
|---|---|---|---|---|---|---|
Harris et al., [169] | 8 | 33 (22 – 42) | 4mg | 12.52 ± 4.37 a | 1.84 ± 0.72 c | 1.06 ± 0.42 |
Predicted | 8 | 30.2 (23 – 43) | 4mg | 12.03 ± 2.91a | 1.65 ± 0.38 c | 1.48 ± 0.33 |
P/O ratio | 0.96 | 0.90 | 1.39 | |||
Harris et al., [169] | 8 | 33 (22 – 42) | 16mg | 35.25 ± 9.89 a | 4.54 ± 1.01 c | 1.51 ± 0.32 |
Predicted | 8 | 30.2 (23 – 43) | 16mg | 32.63 ± 8.23 a | 5.47 ± 1.27 c | 1.04 ± 0.65 |
P/O ratio | 0.93 | 1.20 | 0.68 | |||
Ciraulo et al., [170] | 28 | 33 (21 – 45) | 8mg | 19.92 ± 12.67 b | 2.65 ± 1.05 d | 1.15 ± 0.49 |
Predicted | 28 | 29.6 (22 – 45) | 8mg | 19.61 ± 7.66 b | 2.5 ± 0.83 d | 1.58 ± 0.35 |
P/O ratio | 0.98 | 0.94 | 1.37 | |||
Ciraulo et al., [170] | 28 | 33 (21 – 45) | 24mg | 48.81 ± 31.07 b | 5.41 ± 3.42 d | 0.92 ± 0.45 |
Predicted | 30 (22 – 45) | 24mg | 48.15 ± 22.09 b | 5.87 ± 2.40 d | 1.60 ± 0.37 | |
P/O ratio | 0.98 | 1.08 | 1.74 | |||
Compton et al., [171] | 24 | 48.5 (18 – 65) | 16mg | 70.32 ± 22.64 b | 10.38 ± 3.45 d | 1.24 ± 0.36 |
CV (%) | NR | 33.3 | 29.4 | 32.2 | ||
Minimum | NR | 4.12 | 0.5 | 43.16 | ||
Maximum | NR | 19.51 | 2 | 19.51 | ||
Predicted | 24 | 33.2 (27 – 41) | 16mg | 66.24 ± 20.46 b | 9.27 ± 2.27 d | 1.5 ± 0.41 |
CV (%) | NR | 24 | 28 | 31 | ||
Minimum | NR | 5.96 | 0.9 | 37.91 | ||
Maximum | NR | 13.82 | 2.25 | 109.73 | ||
P/O ratio | 0.94 | 0.89 | 1.20 |
Reported Data | N | Trimester | Dose (mg) | AUC 0-12 ng/ml.hr | Difference % | Cmax ng/ml | Difference % | Tmax (h) | Difference % |
|---|---|---|---|---|---|---|---|---|---|
Bastian et al., [173] | 8 | 2 | 8 | 15.2 ± 1.4a | -27 | 4.0 ± 0.1b | -56 | 1.6 ± 2.8 | -24 |
Predicted | 8 | 2 | 8 | 11.06 ± 2.04 | 1.75 ± 0.28 | 1.22 ± 0.30 | |||
P/O ratio | 0.72 | 0.40 | 0.76 | ||||||
Bastian et al., [173] | 13 | 3 | 10 | 22 ± 1.2a | -40 | 5 ± 0.19b | 58 | 1.0 ± 1.1 | 29 |
Predicted | 13 | 3 | 10 | 13.31 ± 2.75 | 2.10 ± 0.44 | 1.29 ± 0.26 | |||
P/O ratio | 0.61 | 0.42 | 1.29 |
AUC (ng/mL.h) | Tmax (h) | Cmax (ng/mL) | Cmin (ng/mL) | CL (L/h) | CYP3A4 Enzyme Abundancea (pmol P450) | BUP fm CYP3A4 Liver (%) | |
|---|---|---|---|---|---|---|---|
Baseline | 56.66 (17.40) | 1.29(0.31) | 7.69 (2.23) | 0.31(0.18) | 306.72 (88.25) | 6587897.996 (4084787.61) | 34.39 |
GW 5 | 57.18 (14.83) | 1.26 (0.31) | 7.85(1.87) | 0.30 (0.16) | 297.52 (73.87) | 7120473.37 (3769062.41) | 35.30 |
GW 10 | 54.49 (13.57) | 1.24 (0.32) | 7.46 (1.75) | 0.29 (0.15) | 311.09 (75.51) | 7748429.23 (4101456.71) | 36.77 |
GW 15 | 51.85 (12.41)** | 1.21 (0.32) | 7.09 (1.63)** | 0.29 (0.15) | 325.86 (77.50) | 8547025.26 (4524175.56) | 38.54 |
GW 20 | 49.37 (11.39)** | 1.20 (0.31) | 6.74 (1.53)** | 0.29 (0.14) | 341.34 (79.73)** | 9516261.47 (5037218.95) | 40.51 |
GW 25 | 47.09 (10.52)** | 1.19 (0.31) | 6.43 (1.44)** | 0.29 (0.14) | 357.06 (82.14)** | 10656137.87 (5640586.88) | 42.61 |
GW 30 | 45.03 (9.77)** | 1.20 (0.30) | 6.15 (1.37)** | 0.29 (0.13) | 372.68 (84.66)** | 11966654.44 (6334279.35) | 44.78 |
GW 35 | 43.19 (9.15)** | 1.22 (0.30) | 5.90 (1.30)** | 0.29 (0.13) | 387.94 (87.25)** | 13447811.19 (7118296.35) | 46.96 |
GW 40 | 41.56 (8.64)** | 1.27 (0.30) | 5.66 (1.24)** | 0.28 (0.12) | 402.70 (89.90)** | 15099608.11 (7992637.90) | 49.10 |
T1 (Gestational Week 10) T2 (Gestational Week 25) T3 (Gestational Week 35) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
Dose | Phenotype | Cmax (ng/ml) | % > 1 ng/mL | % > 2 ng/mL | Cmax (ng/ml) | % >1 ng/mL | %>22 ng/mL | Cmax (ng/ml) | % > 1 ng/mL | % > 2 ng/mL |
4mg | EM | 1.86 (0.44) | 99 | 0 | 1.61(0.36) | 97 | 0 | 1.47 (0.33) | 92 | 0 |
8mg | EM | 3.73 (0.87) | 100 | 0 | 3.22 (0.72) | 100 | 0 | 2.95 (0.65) | 10 | 0 |
12mg | EM | 5.59 (1.31) | 100 | 0 | 4.82 (1.08) | 100 | 0 | 4.42 (0.98) | 100 | 0 |
16mg | EM | 7.46 (1.75) | 100 | 0 | 6.43 (1.44) | 100 | 0 | 5.90 (1.30) | 100 | 0 |
20mg | EM | 9.32 (2.18) | 100 | 0 | 8.04 (1.80) | 100 | 0 | 7.37 (1.63) | 100 | 0 |
24 mg | EM | 11.19 (2.62) | 100 | 0 | 9.65 (2.16) | 100 | 0 | 8.84 (1.96) | 100 | 0 |
28mg | EM | 13.19 (3.06) | 100 | 1 | 11.26 (2.52) | 100 | 0 | 10.32 (2.28) | 100 | 0 |
32mg | EM | 14.52 (3.49) | 100 | 3 | 12.86 (2.89) | 100 | 0 | 11.79 (2.61) | 100 | 0 |
ADAM | Advanced Dissolution, Absorption and Metabolism |
AUC | Area Under the Curve |
BMT | Buprenorphine Maintenance Therapy |
BUP | Buprenorphine |
BTDS | Buprenorphine Transdermal System |
B3G | Buprenorphine-3-glucuronide |
CAM | Complementary/Alternative Medicine |
CBT | Cognitive Behavioral Therapy |
CYP | Cytochrome P450 |
CYP3A4 | Cytochrome P450 Isoform 3A4 |
CYP2C8 | Cytochrome P450 Isoform 2C8 |
Cmax | Peak Concentration |
Cmin | Trough Concentration |
CAR | Constitutive Androstane Receptor |
CL | Total Clearance |
CLint | Intrinsic Clearance |
CM | Contingency Management |
DDI | Drug-Drug Interaction |
EM | Extensive Metabolizer |
ESM | Electronic Supplementary Material |
F | Bioavailability |
fm | Fraction metabolised |
GABA | Gamma Amino-Butyric Acid |
GW | Gestational Weeks |
HSA | Human Serum Albumin |
HV | Healthy Volunteer |
Indmax | Induction magnitude |
IVIVET | In vitro in Vitro Extrapolation Technique |
ISEF | Intersystem Extrapolation Factor |
Ki | Inhibition Constant |
Kinact | Maximal Inactivation Rate Constant |
KOR | Kappa Opioid Receptor |
Kp | Tissue-partition Coefficient |
MOTHER | Maternal Opioid Treatment Human Experimental Research Study |
MMT | Methadone Maintenance Therapy |
MOR | Mu Opioid Receptor |
NAS | Neonatal Abstinence Syndrome |
N3G | Norbuprenorphine-3-glucuronide |
OUD | Opioid Use Disorder |
OST | Opioid Substitution Therapy |
p-PBPK | pregnancy Physiologically Based Pharmacokinetic Model. |
PBPK | Physiologically Based Pharmacokinetic Modelling |
PK | Pharmacokinetic |
PXR | Pregnane X Receptor |
QT interval | Time Interval Between Onset of Ventricular Depolarization and End of Ventricular Repolarization |
SL | Sublingual |
SIDS | Sudden Infant Death Syndrome |
SPC | Summary of Product Characteristics |
t1/2 | Half-life |
tmax | Time to Maximum Concentration |
T2 | Second Trimester |
T3 | Third Trimester |
TdP | Torsades de Pointes |
UGT | Uridine 5'-diphospho-glucuronosyltransferase Enzymes |
UGT1A1 | Uridine 5'-diphospho-glucuronosyltransferase Enzyme Isoform 1A1 |
UGT1A3 | Uridine 5'-diphospho-glucuronosyltransferase Enzyme Isoform 1A3 |
UGT2B7 | Uridine 5'-diphospho-glucuronosyltransferase Enzyme Isoform 2B7 |
VPC | Visual Predictive Checking |
Vss | Volume of Distribution at Steady-State |
Vd | Volume of Distribution |
Below is the link to the supplementary material:
| [1] | Gossop, M. Living With Drugs. 5th edition. Ashgate Publishing. 1993, p2 |
| [2] |
U.S. Food and Drug Administration. Drug Abuse and Dependence Section of Labeling for Human Prescription Drug and Biological Products. 2019 Available at:
https://www.fda.gov/media/128443/download (Accessed: 7 March 2022). |
| [3] | Ates Bulut, E. and Isik, A. T. Abuse/Misuse of Prescription Medications in Older Adults, Clin. Geriatr. Med. (2022). 38(1): pp. 85–97. |
| [4] | Mcllelan, A. T. Substance Misuse and Substance use Disorders: Why do they Matter in Healthcare? Trans. Am. Clin. Climatol. Assoc. (2017)., 128: pp. 112–130. |
| [5] | Smith, S. M., Dart, R. C., Katz, N. P., Paillard, F., Adams, E. H., Comer, S. D., Degroot, A., Edwards, R. R., Haddox, J. D., Jaffe, J. H., Jones, C. M., Kleber, H. D., Kopecky, E. A., Markman, J. D., Montoya, I. D., O’Brien, C., Roland, C. L., Stanton, M., Strain, E. C., Vorsanger, G., Wasan, A. D., Weiss, R. D., Turk, D. C., Dworkin, R. H.,. Classification and definition of misuse, abuse, and related events in clinical trials: ACTTION systematic review and recommendations. Pain. (2013), 154: 2287–2296. |
| [6] | Herzig, K., Danley, D., Jackson, R., Petersen, R., Chamberlain, L., Gerbert, B.,. Seizing the 9-month moment: addressing behavioral risks in prenatal patients. Patient Educ. Couns. (2006) 61: 228–235. |
| [7] | Ostrea, E. M., Knapp, D. K., Tannenbaum, L., Ostrea, A. R., Romero, A., Salari, V., Ager, J.,. Estimates of illicit drug use during pregnancy by maternal interview, hair analysis, and meconium analysis. J. Pediatr. (2001) 138: 344–348. |
| [8] | Schempf, A. H., Strobino, D. M.,. Drug use and limited prenatal care: an examination of responsible barriers. Am. J. Obstet. Gynecol. (2009) 200: 412. e1–10. |
| [9] |
Substance Abuse and Mental Health Services Administration, S. A. and M. H. S. A.. Substance Abuse and Mental Health Services Administration. (2016) Available at:
https://www.samhsa.gov/data/sites/default/files/NSDUH-DetTabs-2016/NSDUH-DetTabs-2016.pdf |
| [10] | Tackling Drugs Together. Tackling drugs together: a strategy for England. London: HMSO (1995) (Cm.2846). Available at: librarysearch.lse.ac.uk |
| [11] | Havens, J. R., Simmons, L. A., Shannon, L. M., Hansen, W. F.,. Factors associated with substance use during pregnancy: results from a national sample. Drug Alcohol Depend. (2009) 99: 89–95. |
| [12] | Vesga-López, O., Blanco, C., Keyes, K., Olfson, M., Grant, B. F., Hasin, D. S.,. Psychiatric disorders in pregnant and postpartum women in the United States. Arch Gen Psychiatry (2008) 65: 805–815. |
| [13] | Wright, A., Walker, J.,. Management of women who use drugs during pregnancy. Semin. Fetal. Neonatal Med. (2007) 12: 114–118. |
| [14] | Bauer, C. R., Shankaran, S., Bada, H. S., Lester, B., Wright, L. L., Krause-Steinrauf, H., Smeriglio, V. L., Finnegan, L. P., Maza, P. L., Verter, J., The Maternal Lifestyle Study: drug exposure during pregnancy and short-term maternal outcomes. Am. J. Obstet. Gynecol. (2002) 186: 487–495. |
| [15] | Sweeney, P. J., Schwartz, R. M., Mattis, N. G., Vohr, B.,. The effect of integrating substance abuse treatment with prenatal care on birth outcome. J. Perinatol. (2000) 20: 219–224. |
| [16] | El-Mohandes, A., Herman, A. A., Nabil El-Khorazaty, M., Katta, P. S., White, D., Grylack, L.. Prenatal care reduces the impact of illicit drug use on perinatal outcomes. J. Perinatol. (2003) 23: 354–360. |
| [17] | Nutt, D. J., King, L. A., Phillips, L. D., Drug harms in the UK: a multicriteria decision analysis. Lancet (2010) 376: 1558–1565. |
| [18] | Adrian, M., Barry, S. J., Physical and Mental Health Problems Associated with the Use of Alcohol and Drugs. Subst. Use. Misuse. (2003)38: 1575–1614. |
| [19] | Hernandez-Avila, C. A., Rounsaville, B. J., Kranzler, H. R.,. Opioid-, cannabis- and alcohol-dependent women show more rapid progression to substance abuse treatment. Drug Alcohol Depend. (2004) 74: 265–272. |
| [20] | Brienza, R. S., Stein, M. D.,. Alcohol use disorders in primary care: do gender-specific differences exist? J. Gen. Intern. Med. (2002) 17: 387–397. |
| [21] | Swendsen, J., Conway, K. P., Degenhardt, L., Glantz, M., Jin, R., Merikangas, K. R., Sampson, N., Kessler, R. C.,. Mental disorders as risk factors for substance use, abuse and dependence: results from the 10-year follow-up of the National Comorbidity Survey. Addiction (2010) 105: 1117–1128. |
| [22] | Kissin, W. B., Svikis, D. S., Morgan, G. D., Haug, N. A.,. Characterizing pregnant drug-dependent women in treatment and their children. J. Subst. Abuse Treat. (2001)21: 27–34. |
| [23] | Strandberg-Larsen, K., Nielsen, N. R., Grønbaek, M., Andersen, P. K., Olsen, J., Andersen, A.-M. N.,. Binge drinking in pregnancy and risk of fetal death. Obstet. Gynecol. (2008) 111: 602–609. |
| [24] | Lacroix, I., Berrebi, A., Chaumerliac, C., Lapeyre-Mestre, M., Montastruc, J. L., Damase-Michel, C.,. Buprenorphine in pregnant opioid-dependent women: first results of a prospective study. Addiction (2004) 99: 209–214. |
| [25] | Rigg, K. K., Ibañez, G. E.,. Motivations for non-medical prescription drug use: A mixed methods analysis. J. Subst. Abuse Treat. (2010) 39: 236–247. |
| [26] | Waldhoer, M., Bartlett, S. E., Whistler, J. L.,. Opioid receptors. Annu. Rev. Biochem. (2004) 73: 953–990. |
| [27] | Farid, W. O., Dunlop, S. A., Tait, R. J., Hulse, G. K.,. The Effects of Maternally Administered Methadone, Buprenorphine and Naltrexone on Offspring: Review of Human and Animal Data. Curr. Neuropharmacol. (2008) 6: 125–150. |
| [28] | Chahl, L. A. Opioids - mechanisms of action. Aust. Prescr. (1996) 19: 63–65. |
| [29] | Hyman, S. E., Malenka, R. C. Addiction and the brain: the neurobiology of compulsion and its persistence. Nat. Rev. Neurosci. (2001)2: 695–703. |
| [30] | Terenius, L., Johansson, B.,. The opioid systems--panacea and nemesis. Biochem. Biophys. Res.. Commun. (2010)396: 140– 142. |
| [31] | Kalivas, P. W., Volkow, N. D.,. The neural basis of addiction: a pathology of motivation and choice. Am. J. Psychiatry (2005) 162: 1403–1413. |
| [32] | Volkow, N. D., Fowler, J. S., Wang, G.-J., Swanson, J. M., Telang, F.,. Dopamine in Drug Abuse and Addiction: Results of Imaging Studies and Treatment Implications. Arch. Neurol. (2007) 64: 1575–1579. |
| [33] | Binswanger, I. A., Stern, M. F., Deyo, R. A., Heagerty, P. J., Cheadle, A., Elmore, J. G., Koepsell, T. D.,. Release from Prison — A High Risk of Death for Former Inmates. N. Engl. J. Med. (2007)356: 157–165. |
| [34] | Mattick, R. P., Hall, W.,. Are detoxification programmes effective? Lancet (1996) 347: 97–100. |
| [35] | Nestler, E. J.,. Under siege: The brain on opiates. Neuron (1996) 16: 897–900. |
| [36] | Dürsteler-Mac Farland, K. M., Störmer, R., Seifritz, E., Hug, I., Müller-Spahn, F., Ladewig, D., Stohler, R.,. Opioid- associated effects on oxygen saturation. Addiction (2000) 95: 285–287. |
| [37] | Hulse, G. K., English, D. R., Milne, E., Holman, C. D.,. The quantification of mortality resulting from the regular use of illicit opiates. Addiction (1999) 94: 221–229. |
| [38] | Marsch, L. A. The efficacy of methadone maintenance interventions in reducing illicit opiate use, HIV risk behavior and criminality: a meta-analysis. Addiction (1998) 93: 515–532. |
| [39] | Crome, I. B., Kumar, M. T.,. Epidemiology of drug and alcohol use in young women. Semin. Fetal. Neonatal. Med (2007) 12: 98–105. |
| [40] | Lester, B. M., ElSohly, M., Wright, L. L., Smeriglio, V. L., Verter, J., Bauer, C. R., Shankaran, S., Bada, H. S., Walls, H. H., Huestis, M. A., Finnegan, L. P., Maza, P. L.,. The Maternal Lifestyle Study: drug use by meconium toxicology and maternal self-report. Pediatrics (2001)107: 309–317. |
| [41] | Hulse, G. K., Milne, E., English, D. R., Holman, C. D.,. The relationship between maternal use of heroin and methadone and infant birth weight. Addiction (1997) 92: 1571–1579. |
| [42] | Hulse, G. K., Milne, E., English, D. R., Holman, C. D.,. Assessing the relationship between maternal opiate use and antepartum haemorrhage. Addiction (1998a) 93: 1553–1558. |
| [43] | Hulse, G. K., Milne, E., English, D. R., Holman, C. D.,. Assessing the relationship between maternal opiate use and neonatal mortality. Addiction (1998b) 93: 1033–1042. |
| [44] | Gillogley, K. M., Evans, A. T., Hansen, R. L., Samuels, S. J., Batra, K. K.,. The perinatal impact of cocaine, amphetamine, and opiate use detected by universal intrapartum screening. Am. J Obstet. Gynecol. (1990)163: 1535–1542. |
| [45] | Chasnoff, I. J., Hatcher, R., Burns, W. J.,. Polydrug- and methadone-addicted newborns: a continuum of impairment? Pediatrics (1982) 70: 210–213. |
| [46] | Unger, A. S., Martin, P. R., Kaltenbach, K., Stine, S. M., Heil, S. H., Jones, H. E., Arria, A. M., Coyle, M. G., Selby, P., Fischer, G.,. Clinical characteristics of central European and North American samples of pregnant women screened for opioid agonist treatment. Eur. Addict Res. (2010) 16: 99–107. |
| [47] | Santolaria-Fernández, F. J., Gómez-Sirvent, J. L., González-Reimers, C. E., Batista-López, J. N., Jorge-Hernández, J. A., Rodríguez-Moreno, F., Martínez-Riera, A., Hernández-García, M. T.,. Nutritional assessment of drug addicts. Drug Alcohol Depend. (1995) 38: 11–18. |
| [48] | Jansson, L. M., Svikis, D., Lee, J., Paluzzi, P., Rutigliano, P., Hackerman, F.. Pregnancy and addiction A comprehensive care model. J. Subst. Abuse Treat. (1996) 13: 321–329. |
| [49] | Kennare, R., Heard, A., Chan, A. Substance use during pregnancy: risk factors and obstetric and perinatal outcomes in South Australia. Aust. N. Z. J. Obstet. Gynaecol. (2005) 45: 220–225. |
| [50] | Luo, Z.-C., Wilkins, R., Kramer, M. S.,. Effect of neighbourhood income and maternal education on birth outcomes: a population-based study. CMAJ (2006) 174(10): 1415–1420. |
| [51] | Myllynen, P., Pasanen, M., Pelkonen, O. Human placenta: a human organ for developmental toxicology research and biomonitoring. Placenta (2005) 26: 361–371. |
| [52] | Myren, M., Mose, T., Mathiesen, L., Knudsen, L. E.. The human placenta – An alternative for studying foetal exposure., Fourteenth International Workshop on In Vitro Toxicology. Toxicol. In Vitro (2007) 21: 1332–1340. |
| [53] | Gareri, J., Klein, J., Koren, G.. Drugs of abuse testing in meconium. Clin. Chim. Acta. (2006) 366: 101–111. |
| [54] | Levitt, P., Prenatal effects of drugs of abuse on brain development. Drug Alcohol Depend. (1998) 51: 109–125. |
| [55] | Farrell, T., Owen, P., Harrold, A.,. Fetal movements following intrapartum maternal opiate administration. Clin Exp Obstet. Gynecol. (1996) 23: 144–146. |
| [56] | Wouldes, T. A., Roberts, A. B., Pryor, J. E., Bagnall, C., Gunn, T. R.,. The effect of methadone treatment on the quantity and quality of human fetal movement. Neurotoxicol. Teratol. (2004) 26: 23–34. |
| [57] | Navaneethakrishnan, R., Tutty, S., Sinha, C., Lindow, S. W.,. The effect of maternal methadone use on the fetal heart pattern: a computerised CTG analysis. BJOG (2006) 113: 948–950. |
| [58] | Ney, J. A., Dooley, S. L., Keith, L. G., Chasnoff, I. J., Socol, M. L.,. The prevalence of substance abuse in patients with suspected preterm labor. Am. J. Obstet. Gynecol. (1990) 162: 1562–1565 |
| [59] | Pinto, S. M., Dodd, S., Walkinshaw, S. A., Siney, C., Kakkar, P., Mousa, H. A.,. Substance abuse during pregnancy: effect on pregnancy outcomes. Eur. J. Obstet. Gynecol. Reprod. Biol. (2010) 150: 137–141. |
| [60] | Coyle, M. G., Brogly, S. B., Ahmed, M. S., Patrick, S. W., Jones, H. E., Neonatal abstinence syndrome. Nat Rev Dis Primers (2018) 4: 47. |
| [61] | Mégarbane, B., Marie, N., Pirnay, S., Borron, S. W., Gueye, P. N., Risède, P., Monier, C., Noble, F., Baud, F. J.,. Buprenorphine is protective against the depressive effects of norbuprenorphine on ventilation. Toxicol. Appl. Pharmacol. (2006) 212: 256–267. |
| [62] | Lintzeris, N.,. Prescription of heroin for the management of heroin dependence: current status. CNS Drugs. (2009) 23: 463– 476. |
| [63] | Thorngren-Jerneck, K., Herbst, A.,. Low 5-minute Apgar score: a population-based register study of 1 million term births. Obstet. Gynecol. (2001) 98: 65–70. |
| [64] | Volmanen, P., Sarvela, J., Akural, E. I., Raudaskoski, T., Korttila, K., Alahuhta, S.,. Intravenous remifentanil vs. epidural levobupivacaine with fentanyl for pain relief in early labour: a randomised, controlled, double-blinded study. Acta. Anaesthesiol. Scand. (2008) 52: 249–255. |
| [65] | Connaughton, J. F., Reeser, D., Schut, J., Finnegan, L. P.,. Perinatal addiction: Outcome and management. Am J Obstet. Gynecol. (1977) 129: 679–686. |
| [66] | Kaltenbach, K., Berghella, V., Finnegan, L.,. Opioid dependence during pregnancy. Effects and management. Obstet Gynecol. Clin. North Am. (1998) 25: 139–151. |
| [67] | Kraft, W. K., Dysart, K., Greenspan, J. S., Gibson, E., Kaltenbach, K., Ehrlich, M. E.,. Revised dose schema of sublingual buprenorphine in the treatment of the neonatal opioid abstinence syndrome. Addiction (2011) 106: 574–580. |
| [68] | Bada, H., Bauer, C., Shankaran, S., Lester, B., Wright, L., Das, A., Poole, K., Smeriglio, V., Finnegan, L., Maza, P.,. Central and autonomic system signs with in utero drug exposure. Arch. Dis. Child Fetal. Neonatal. Ed. (2002) 87: F106–F112. |
| [69] | Finnegan, L. P., Connaughton, J. F., Kron, R. E., Emich, J. P.,. Neonatal abstinence syndrome: assessment and management. Addict Dis (1975a) 2: 141–158. |
| [70] | Kakko, J., Heilig, M., Sarman, I., Buprenorphine and methadone treatment of opiate dependence during pregnancy: comparison of fetal growth and neonatal outcomes in two consecutive case series. Drug Alcohol Depend. (2008) 96: 69–78. |
| [71] | Ebner, N., Rohrmeister, K., Winklbaur, B., Baewert, A., Jagsch, R., Peternell, A., Thau, K., Fischer, G., Management of neonatal abstinence syndrome in neonates born to opioid maintained women. Drug Alcohol Depend. (2007). 87: 131–138. |
| [72] | Langenfeld, S., Birkenfeld, L., Herkenrath, P., Müller, C., Hellmich, M., Theisohn, M., Therapy of the neonatal abstinence syndrome with tincture of opium or morphine drops. Drug Alcohol Depend. (2005). 77: 31–36. |
| [73] | Coyle, M. G., Ferguson, A., Lagasse, L., Oh, W., Lester, B., Diluted tincture of opium (DTO) and phenobarbital versus DTO alone for neonatal opiate withdrawal in term infants. J. Pediatr. (2002) 140: 561–564. |
| [74] | Ornoy, A., Segal, J., Bar-Hamburger, R., Greenbaum, C., Developmental outcome of school-age children born to mothers with heroin dependency: importance of environmental factors. Dev. Med. Child Neurol. (2001) 43: 668–675. |
| [75] | Kahlert, C., Rudin, C., Kind, C., (SHCS), and the S. H. C. S., Sudden infant death syndrome in infants born to HIV‐ infected and opiate‐using mothers. Arch. Dis. Child (2007). 92: 1005–1008. |
| [76] | Kandall, S. R., Doberczak, T. M., Jantunen, M., Stein, J.,. The methadone-maintained pregnancy. Clin. Perinatol. (1999) 26: 173–183. |
| [77] | Hunt, C. E., Hauck, F. R. Sudden infant death syndrome. CMAJ (2006) 174: 1861–1869. |
| [78] | Suguihara, C., Bancalari, E., Substance abuse during pregnancy: effects on respiratory function in the infant. Semin. Perinatol. (1991). 15: 302–309. |
| [79] | Connock, M., Juarez-Garcia, A., Jowett, S., Frew, E., Liu, Z., Taylor, R. J., Fry-Smith, A., Day, E., Lintzeris, N., Roberts, T., Burls, A., Taylor, R. S., Methadone and buprenorphine for the management of opioid dependence: a systematic review and economic evaluation. Health Technol. Assess (2007). 11: 1–171, iii–iv. |
| [80] |
Miller, W. R. and Rollnick, S. Motivational interviewing: Preparing people to change addictive behavior. New York: Guilford Press. (1991). Available at:
https://onlinelibrary.wiley.com/doi/abs/10.1002/casp.2450020410 (Accessed: 8 March 2022). |
| [81] | Beck, A. T. Cognitive therapy and the emotional disorders. Oxford, England: International Universities Press, (1976) p. 356. |
| [82] | Ernst, E., Resch, K. L., Mills, S., Hill, R., Mitchell, A., Willoughby, M., White, A.,. Complementary medicine — a definition. Br. J. Gen. Pract. (1995) 45: 506. |
| [83] | Beck, A. T., Wright, F. D., Newman, C. F., Liese, B.,. Cognitive Therapy of Substance Abuse. (2001). |
| [84] | Rementeriá, J. L., Nunag, N. N., (1973). Narcotic withdrawal in pregnancy: stillbirth incidence with a case report. Am. J. Obstet. Gynecol. 116: 1152–1156. |
| [85] | Zuspan, F. P., Gumpel, J. A., Mejia-Zelaya, A., Madden, J., Davis, R., (1975). Fetal stress from methadone withdrawal. Am. J. Obstet. Gynecol. 122: 43–46. |
| [86] | Dashe, J. S., Jackson, G. L., Olscher, D. A., Zane, E. H., Wendel, G. D., (1998). Opioid detoxification in pregnancy. Obstet Gynecol 92: 854–858. |
| [87] | Luty, J., Nikolaou, V., Bearn, J., (2003). Is opiate detoxification unsafe in pregnancy? J. Subst. Abuse Treat. 24: 363–367. |
| [88] | Farrell, M., Ward, J., Mattick, R., Hall, W., Stimson, G. V., Jarlais, D. des, Gossop, M., Strang, J., (1994). Fortnightly Review: Methadone maintenance treatment in opiate dependence: a review. BMJ 309: 997–1001. |
| [89] | Ward, J., Hall, W., Mattick, R. P., Role of maintenance treatment in opioid dependence. Lancet (1999a) 353: 221–226. |
| [90] | Rayburn, W. F., Bogenschutz, M. P., Pharmacotherapy for pregnant women with addictions. Am. J. Obstet. Gynecol. 2004a, 191: 1885–1897. |
| [91] | Fiellin, D. A., Pantalon, M. V., Chawarski, M. C., Moore, B. A., Sullivan, L. E., O’Connor, P. G., Schottenfeld, R. S. Counseling plus buprenorphine-naloxone maintenance therapy for opioid dependence. N Engl. J. Med., (2006) 355: 365–374. |
| [92] | McLellan, A. T., Arndt, I. O., Metzger, D. S., Woody, G. E., O’Brien, C. P., The effects of psychosocial services in substance abuse treatment. JAMA,1993 269: 1953–1959. |
| [93] | Public Health England. NDTMS (national drug treatment monitoring system). Public Health England. (2021). Retrieved 29th July from |
| [94] | Fudala, P. J., Bridge, T. P., Herbert, S., Williford, W. O., Chiang, C. N., Jones, K., Collins, J., Raisch, D., Casadonte, P., Goldsmith, R. J., Ling, W., Malkerneker, U., McNicholas, L., Renner, J., Stine, S., Tusel, D., Office-based treatment of opiate addiction with a sublingual-tablet formulation of buprenorphine and naloxone. N Engl. J Med. (2003) 349: 949–958. |
| [95] | Yaksh, T., Wallace, M.. Chapter 20 - Opioids, Analgesia, and Pain Management, in: Brunton, L. L., Knollmann, B. C., Hilal-Dandan, R. (Eds.), Goodman & Gilman’s: The Pharmacological Basis of Therapeutics. McGraw Hill Medical, New York, (2018) pp. 355–386. |
| [96] | Khanna, I. K., Pillarisetti, S. Buprenorphine – an attractive opioid with underutilized potential in treatment of chronic pain. J. Pain Res. (2015) 8: 859–870. |
| [97] | Tröster, A., Ihmsen, H., Singler, B., Filitz, J., Koppert, W. Interaction of fentanyl and buprenorphine in an experimental model of pain and central sensitization in human volunteers. Clin. J. Pain (2012) 28: 705–711. |
| [98] | Whelan, P. J. and Remski, K. Buprenorphine vs methadone treatment: A review of evidence in both developed and developing worlds, J. Neurosci. Rural Pract. (2012) 3(1): pp. 45–50. |
| [99] | Auriacombe, M., Fatséas, M., Dubernet, J., Daulouède, J.-P., Tignol, J., French field experience with buprenorphine. Am. J. Addict. (2004). 13(1): S17-28. |
| [100] | Fischer, G., Ortner, R., Rohrmeister, K., Jagsch, R., Baewert, A., Langer, M., Aschauer, H., Methadone versus buprenorphine in pregnant addicts: a double-blind, double-dummy comparison study. Addiction (2006) 101: 275–281. |
| [101] | Dole, V. P., Nyswander, M. E., Kreek, M. J. Narcotic blockade. Arch. Intern. Med (1966) 118: 304–309. |
| [102] | Dole, V. P., Nyswander, M., A Medical Treatment for Diacetylmorphine (Heroin) Addiction: A Clinical Trial With Methadone Hydrochloride. JAMA (1965). 193: 646–650. |
| [103] | Sees, K. L., Delucchi, K. L., Masson, C., Rosen, A., Clark, H. W., Robillard, H., Banys, P., Hall, S. M. Methadone maintenance vs 180-day psychosocially enriched detoxification for treatment of opioid dependence: a randomized controlled trial. JAMA (2000) 283: 1303–1310. |
| [104] | Gruber, V. A., Delucchi, K. L., Kielstein, A., Batki, S. L. A randomized trial of 6-month methadone maintenance with standard or minimal counseling versus 21-day methadone detoxification. Drug. Alcohol. Depend. (2008). 94: 199–206. |
| [105] | Cairns, A., Roberts, I. S., Benbow, E. W., Characteristics of fatal methadone overdose in Manchester, 1985-94. BMJ 1996, 313: 264–265. |
| [106] | Krantz, M. J., Kutinsky, I. B., Robertson, A. D., Mehler, P. S., Dose-related effects of methadone on QT prolongation in a series of patients with torsade de pointes. Pharmacotherapy (2003). 23: 802–805. |
| [107] | Fonseca, F., Marti-Almor, J., Pastor, A., Cladellas, M., Farré, M., de la Torre, R., Torrens, M., Prevalence of long QTc interval in methadone maintenance patients. Drug Alcohol Depend. (2009). 99: 327–332. https://doi.org/10.1016/j.drugalcdep.2008.06.018 |
| [108] | Strain, E. C., Bigelow, G. E., Liebson, I. A., Stitzer, M. L., Moderate- vs high-dose methadone in the treatment of opioid dependence: a randomized trial. JAMA (1999). 281: 1000–1005. |
| [109] | Burns, L., Mattick, R. P., Lim, K., Wallace, C., Methadone in pregnancy: treatment retention and neonatal outcomes. Addiction (2007). 102: 264–270. |
| [110] | Elkader, A. and Sproule, B. Buprenorphine, Clin. Pharmacokinet (2005) 44(7): pp. 661–680. |
| [111] | Kuhlman, J. J., Jr., Lalani, S., Magluilo, J., Jr., Levine, B., Darwin, W. D., Johnson, R. E., Cone, E. J., Human Pharmacokinetics of Intravenous, Sublingual, and Buccal Buprenorphine*. J. Anal. Toxicol. (1996). 20: 369–378. |
| [112] | Marquet, P., Pharmacology of High-Dose Buprenorphine, in: Kintz, P. (Ed.), Buprenorphine Therapy of Opiate Addiction, Forensic Science and Medicine. Humana Press, Totowa, NJ, (2002). pp. 1–11. |
| [113] | Saleem, B., Conaghan, P. G., Chapter 15 - Pharmacological treatments in rheumatic diseases, in: Dziedzic, K., Hammond, A. (Eds.), Rheumatology. Churchill Livingstone, Edinburgh, pp. (2010). 199–209. |
| [114] | Davis, M. P. Buprenorphine in cancer pain. Support Care Cancer, (2005) 13(11): pp. 878–887. |
| [115] | Pande, L., Piper, B., An Examination of the Complex Pharmacological Properties of the Non-Selective Opioid Receptor Modulator Buprenorphine. (2020). |
| [116] | Gudin, J., Fudin, J., A Narrative Pharmacological Review of Buprenorphine: A Unique Opioid for the Treatment of Chronic Pain. Pain. Ther. (2020). 9: 41–54. |
| [117] |
Specialist Pharmacists in Substance Abuse. Guidance for Use of Buprenorphine Products for the Treatment of Opioid Dependence in NHS Grampian. (2019) Available at:
https://www.nhsgrampian.org/globalassets/foidocument/foi-public- documents1---all-documents/Guide_Buprenorphine.pdf (Accessed: 9 March 2022). |
| [118] | Wallace, L., Kadakia, A. Buprenorphine transdermal system utilization, Postgrad. Med. J (2017). 129(1): pp. 81–86. |
| [119] | Chavoustie, S., Frost, M., Snyder, O., Owen, J., Darwish, M., Dammerman, R., Sanjurjo, V., Buprenorphine implants in medical treatment of opioid addiction. Expert Rev. Clin. Pharmacol (2017). 10: 799–807. |
| [120] | Packhaeuser, C. B., Schnieders, J., Oster, C. G., Kissel, T., In situ forming parenteral drug delivery systems: an overview. Eur. J. Pharm. Biopharm. (2004). 58: 445–455. |
| [121] | Nasser, A. F., Heidbreder, C., Gomeni, R., Fudala, P. J., Zheng, B., Greenwald, M. K., A Population Pharmacokinetic and Pharmacodynamic Modelling Approach to Support the Clinical Development of RBP-6000, a New, Subcutaneously Injectable, Long-Acting, Sustained-Release Formulation of Buprenorphine, for the Treatment of Opioid Dependence. Clin. Pharmacokinet. (2014). 53: 813–824. |
| [122] | Parry, E., Shields, R., and Turnbull, A. C. Transit Time in the Small Intestine in Pregnancy. Bjog Bjog-Int J Obstet Gy, (1970) (Suppl 10) 77: pp. 900–901. |
| [123] | Dawes, M. and Chowienczyk, P. J. Pharmacokinetics in pregnancy. Best Pract. Res. Clin. Obstet. Gynaecol. (2001) 15(6): pp. 819–826. |
| [124] | Clements, J. A., Heading, R. C., Nimmo, W. S., Prescott, L. F. Kinetics of acetaminophen absorption and gastric emptying in man. Clin. Pharm. Therap. (1978). 24: 420–431. |
| [125] | Costantine, M., Physiologic and pharmacokinetic changes in pregnancy. Front. Pharmacol. (2014). 5: |
| [126] | Cheung, C. K., Lao, T., Swaminathan, R., Urinary excretion of some proteins and enzymes during normal pregnancy. Clin Chem. (1989). 35: 1978–1980. |
| [127] | Erman, A., Neri, A., Sharoni, R., Rabinov, M., Kaplan, B., Rosenfeld, J. B., Boner, G., Enhanced urinary albumin excretion after 35 weeks of gestation and during labour in normal pregnancy. Scand. J. Clin. Lab. Invest. (1992). 52: 409–413. |
| [128] | Hayashi, M., Ueda, Y., Hoshimoto, K., Ota, Y., Fukasawa, I., Sumori, K., Kaneko, I., Abe, S., Uno, M., Ohkura, T., Inaba, N., Changes in urinary excretion of six biochemical parameters in normotensive pregnancy and preeclampsia. Am. J. Kidney Dis. (2002). 39: 392–400. |
| [129] | Feghali, M., Venkataramanan, R. and Caritis, S. Pharmacokinetics of drugs in pregnancy. Semin. Perinatol. (2015). 39(7): pp. 512–519. |
| [130] | Davidson, J. M and Dunlop, W. Changes in renal hemodynamics and tubular function induced by normal human pregnancy., Semi. Nephrol., (1984). 4: pp. 198–207. |
| [131] | Barron, W. M. and Lindheimer, M. D. Renal sodium and water handling in pregnancy. Obstet. Gynecol. (1984). 13: pp. 35–69. |
| [132] | Hutchings, D. E., Hamowy, A. S., Williams, E. M., Zmitrovich, A. C., Prenatal administration of buprenorphine in the rat: Effects on the rest-activity cycle at 22 and 30 days of age. Pharmacol. Biochem. Behav. (1996). 55: 607–613. |
| [133] | Anderson, G. D. Pregnancy-Induced Changes in Pharmacokinetics. Clin. Pharmacokinet., (2005). 44(10): pp. 989–1008. |
| [134] | Selvi, U. P. G., Kamatchi, D., Jeyashri, S., Chanthinidevi, A., Prevalence of Oral Lesions and Measurement of Salivary pH in the Different Trimesters of Pregnancy. Int. J. Sci. Study. (2017). 4: 164–168. |
| [135] | Lacroix, I., Berrebi, A., Garipuy, D., Schmitt, L., Hammou, Y., Chaumerliac, C., Lapeyre-Mestre, M., Montastruc, J.-L., Damase-Michel, C., Buprenorphine versus methadone in pregnant opioid-dependent women: a prospective multicenter study. Eur. J. Clin. Pharmacol. (2011). 67: 1053. |
| [136] | Hytinantti, T., Kahila, H., Renlund, M., Järvenpää, A.-L., Halmesmäki, E., Kivitie-Kallio, S., Neonatal outcome of 58 infants exposed to maternal buprenorphine in utero. Acta. Paediatrica. (2008). 97: 1040–1044. |
| [137] | Jones, H. E., Kaltenbach, K., Heil, S. H., Stine, S. M., Coyle, M. G., Arria, A. M., O’Grady, K. E., Selby, P., Martin, P. R., Fischer, G., Neonatal abstinence syndrome after methadone or buprenorphine exposure. N. Engl. J. Med. (2010). 363: 2320–2331. |
| [138] | Welle-Strand, G. K., Skurtveit, S., Jones, H.E., Waal, H., Bakstad, B., Bjarkø, L., Ravndal, E., Neonatal outcomes following in utero exposure to methadone or buprenorphine: A National Cohort Study of opioid-agonist treatment of Pregnant Women in Norway from 1996 to 2009. Drug Alcohol Depend. (2013). 127: 200–206. |
| [139] | Alsmadi, M. M., Salivary Therapeutic Monitoring of Buprenorphine in Neonates After Maternal Sublingual Dosing Guided by Physiologically Based Pharmacokinetic Modeling. Ther Drug Monit 2024. 46, 512–521. |
| [140] | Bullingham, R. E., McQuay, H. J., Dwyer, D., Allen, M. C., Moore, R. A., Sublingual buprenorphine used postoperatively: clinical observations and preliminary pharmacokinetic analysis. Br J Clin Pharmacol 1981. 12, 117–122. |
| [141] | Caritis, S. N., Bastian, J. R., Zhang, H., Kalluri, H., English, D., England, M., Bobby, S., Venkataramanan, R., An evidence-based recommendation to increase the dosing frequency of buprenorphine during pregnancy. Am J Obstet Gynecol 2017. 217, 459. e1-459. e6. |
| [142] | Eudy-Byrne, R., Zane, N., Adeniyi-Jones, S. C., Gastonguay, M. R., Ruiz-Garcia, A., Kaushal, G., Kraft, W. K., Pharmacometric dose optimization of buprenorphine in neonatal opioid withdrawal syndrome. Clin Transl Sci, 2021. 14, 2171–2183. |
| [143] | Shenkoya, B., Gopalakrishnan, M., Eke, A. C., Physiologically based pharmacokinetic modeling of long-acting extended-release naltrexone in pregnant women with opioid use disorder. CPT Pharmacometrics Syst Pharmacol. 2024. |
| [144] | van Hoogdalem, M. W., Johnson, T. N., McPhail, B. T., Kamatkar, S., Wexelblatt, S. L., Ward, L. P., Christians, U., Akinbi, H. T., Vinks, A. A., Mizuno, T., Physiologically-Based Pharmacokinetic Modeling to Investigate the Effect of Maturation on Buprenorphine Pharmacokinetics in Newborns with Neonatal Opioid Withdrawal Syndrome. Clin Pharmacol Ther 2022a.111, 496–508. |
| [145] | van Hoogdalem, M. W., Tanaka, R., Abduljalil, K., Johnson, T. N., Wexelblatt, S. L., Akinbi, H. T., Vinks, A. A., Mizuno, T., Forecasting Fetal Buprenorphine Exposure through Maternal-Fetal Physiologically Based Pharmacokinetic Modeling. Pharmaceutics 2024a. 16, 375. |
| [146] | van Hoogdalem, M. W., Tanaka, R., Johnson, T. N., Vinks, A. A., Mizuno, T., Development and Verification of a Full Physiologically Based Pharmacokinetic Model for Sublingual Buprenorphine in Healthy Adult Volunteers that Accounts for Nonlinear Bioavailability. Drug Metab Dispos 2024b. 52, 785–796. |
| [147] | van Hoogdalem, M. W., Wexelblatt, S. L., Akinbi, H. T., Vinks, A. A., Mizuno, T., A review of pregnancy-induced changes in opioid pharmacokinetics, placental transfer, and fetal exposure: Towards fetomaternal physiologically-based pharmacokinetic modeling to improve the treatment of neonatal opioid withdrawal syndrome. Pharmacol Ther 2022b. 234, 108045. |
| [148] | Walsh, S. L., Preston, K. L., Stitzer, M. L., Cone, E. J., Bigelow, G. E., Clinical pharmacology of buprenorphine: ceiling effects at high doses. Clin Pharmacol Ther 1994. 55, 569–580. |
| [149] | Zhang, H., Bastian, J. R., Zhao, W., Chen, H., Shaik, I. H., Chaphekar, N., Caritis, S. N., Venkataramanan, R., Pregnancy Alters CYP- and UGT-Mediated Metabolism of Buprenorphine. Ther Drug Monit 2020. 42, 264–270. |
| [150] | Zhang, H., Kalluri, H. V., Bastian, J. R., Chen, H., Alshabi, A., Caritis, S. N., Venkataramanan, R., Gestational changes in buprenorphine exposure: A physiologically-based pharmacokinetic analysis. Br J Clin Pharmacol 2018. 84, 2075–2087. |
| [151] | Sager, J. E., Yu, J., Ragueneau-Majlessi, I., Isoherranen, N., Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulation Approaches: A Systematic Review of Published Models, Applications, and Model Verification. Drug Metab. Dispos. (2015). 43: 1823–1837. |
| [152] |
Ke, A., Dosing for Two: How Pharmacometrics Supports Drug Safety in Pregnancy. Certara. (2015). URL
https://www.certara.com/blog/dosing-for-two-how-pharmacometrics-supports-drug-safety-in-pregnancy/ (accessed 3.24.22). |
| [153] | Abduljalil, K., Furness, P., Johnson, T. N., Rostami-Hodjegan, A., Soltani, H., Anatomical, physiological and metabolic changes with gestational age during normal pregnancy: a database for parameters required in physiologically based pharmacokinetic modelling. Clin. Pharmacokinet. (2012). 51: 365–396. |
| [154] | Gaohua, L., Abduljalil, K., Jamei, M., Johnson, T. N., Rostami-Hodjegan, A., A pregnancy physiologically based pharmacokinetic (p-PBPK) model for disposition of drugs metabolized by CYP1A2, CYP2D6 and CYP3A4. Br. J Clin. Pharmacol. (2012). 74: 873–885. |
| [155] | Rowland Yeo, K., Jamei, M., Yang, J., Tucker, G. T., Rostami-Hodjegan, A., Physiologically based mechanistic modelling to predict complex drug-drug interactions involving simultaneous competitive and time-dependent enzyme inhibition by parent compound and its metabolite in both liver and gut - the effect of diltiazem on the time-course of exposure to triazolam. Eur. J. Pharm. Sci. (2010). 39: 298–309. |
| [156] | De Sousa Mendes, M., Hirt, D., Urien, S., Valade, E., Bouazza, N., Foissac, F., Blanche, S., Treluyer, J.-M., Benaboud, S., Physiologically-based pharmacokinetic modeling of renally excreted antiretroviral drugs in pregnant women. Br. J Clin. Pharmacol (2015). 80: 1031–1041. |
| [157] | Lu, G., Abduljalil, K., Jamei, M., Johnson, T. N., Soltani, H., Rostami-Hodjegan, A., Physiologically-based pharmacokinetic (PBPK) models for assessing the kinetics of xenobiotics during pregnancy: achievements and shortcomings. Curr. Drug. Metab. (2012). 13: 695–720. |
| [158] | Jogiraju, V. K., Avvari, S., Gollen, R., Taft, D. R., Application of physiologically based pharmacokinetic modeling to predict drug disposition in pregnant populations. Biopharm. Drug Dispos. (2017). 38: 426–438. |
| [159] | Olafuyi, O., Badhan, R. K. S., Dose Optimization of Chloroquine by Pharmacokinetic Modeling During Pregnancy for the Treatment of Zika Virus Infection. J. Pharm. Sci. (2019). 108: 661–673. |
| [160] | Bai SA, Xiang Q, Finn A Evaluation of the Pharmacokinetics of Single- and Multiple-dose Buprenorphine Buccal Film in Healthy Volunteers. Clinical Therapeutics (2016) 38(2): 358–369. |
| [161] | Mendelson, J., Upton, R. A., Everhart, E. T., Jacob, P., Jones, R. T.,. Bioavailability of sublingual buprenorphine. J. Clin. Pharmacol. (1997) 37: 31–37. |
| [162] | Bullingham RE, McQuay HJ, Moore A, Bennett MR Buprenorphine kinetics. Clinical pharmacology and therapeutics (1980) 28(5): 667–72. |
| [163] | Bartlett AJ, Lloyd-Jones JG, Rance MJ, Flockhart IR, Dockray GJ, Bennett MR, Moore RA The radioimmunoassay of buprenorphine. European Journal of Clinical Pharmacology (1980) 18(4): 339–345. |
| [164] | Bullingham RE, McQuay HJ, Porter EJ, Allen MC, Moore RA. Sublingual buprenorphine used postoperatively: ten hour plasma drug concentration analysis. British journal of clinical pharmacology (1982) 13(5): 665–73. |
| [165] | Huestis M, Cone E, Pirnay S, Umbricht A, Preston K. Intravenous buprenorphine and norbuprenorphine pharmacokinetics in humans. Drug and Alcohol Dependence (2013) 131(3): 258–262. |
| [166] | Harris, D. S.; Jones, R. T.; Welm, S.; Upton, R. A.; Lin, E.; Mendelson, J. Buprenorphine and naloxone co-administration in opiate-dependent patients stabilized on sublingual buprenorphine. Drug Alcohol Depend. 2000, 61, 85–94. |
| [167] | Lim SCB, Schug S, and Krishnarajah J. The pharmacokinetics and local tolerability of a novel sublingual formulation of buprenorphine. Pain Med (2019) 20: 143-152. |
| [168] | Wojtyniak, J.-G., Britz, H., Selzer, D., Schwab, M., Lehr, T., Data Digitizing: Accurate and Precise Data Extraction for Quantitative Systems Pharmacology and Physiologically-Based Pharmacokinetic Modeling. CPT Pharmacometrics Syst Pharmacol 2020. 9, 322–331. |
| [169] | Harris, D. S., Mendelson, J. E., Lin, E. T., Upton, R. A., Jones, R. T., Pharmacokinetics and Subjective Effects of Sublingual Buprenorphine, Alone or in Combination with Naloxone. Clin. Pharmacokinet. (2004). 43: 329–340. |
| [170] | Ciraulo, D. A., Hitzemann, R. J., Somoza, E., Knapp, C. M., Rotrosen, J., Sarid-Segal, O., Ciraulo, A. M., Greenblatt, D. J., Chiang, C. N., Pharmacokinetics and Pharmacodynamics of Multiple Sublingual Buprenorphine Tablets in Dose-Escalation Trials. J. Clin. Pharmacol. (2006). 46: 179–192. |
| [171] | Compton, P., Ling, W., Moody, D., Chiang, N.,. Pharmacokinetics, bioavailability and opioid effects of liquid versus tablet buprenorphine. Drug Alcohol Depend. (2006) 82: 25–31. |
| [172] | Rescigno, A., Beck, J. S., Thakur, A. K.,. The use and abuse of models. J. Pharmacokinet. Pharmacodyn. (1987) 15: 327–340. |
| [173] | Bastian, J. R., Chen, H., Zhang, H., Rothenberger, S., Tarter, R., English, D., Venkataramanan, R., Caritis, S. N., Dose- adjusted plasma concentrations of sublingual buprenorphine are lower during than after pregnancy. Am. J. Obstet. Gynecol. (2017). 216: 64. e1-64. e7. |
| [174] | Kalluri, H. V., Zhang, H., Caritis, S. N., Venkataramanan, R., A physiologically based pharmacokinetic modelling approach to predict buprenorphine pharmacokinetics following intravenous and sublingual administration. Br. J. Clin. Pharmacol. (2017). 83: 2458–2473. |
| [175] | Almurjan, A., Macfarlane, H., Badhan, R. K. S., Precision dosing-based optimisation of paroxetine during pregnancy for poor and ultrarapid CYP2D6 metabolisers: a virtual clinical trial pharmacokinetics study. J. Pharm. Pharmacol. (2020). 72: 1049–1060. |
| [176] |
U.S. Food and Drug Administration. Draft Guidance for Industry: Drug Interaction Studies--Study Design, Data Analysis, Implications for Dosing, and Labeling Recommendations. U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER). 2012. Available at:
https://downloads.regulations.gov/FDA-2006-D-0036-0032/content.pdf (Accessed: 3 August 2022). |
| [177] | Greenwald, M., Johanson, C.-E., Bueller, J., Chang, Y., Moody, D. E., Kilbourn, M., Koeppe, R., Zubieta, J.-K., Buprenorphine Duration of Action: Mu-opioid Receptor Availability and Pharmacokinetic and Behavioral Indices. Biol. Psychiatry (2007). 61: 101–110. |
| [178] | Nahar, L. K., Andrews, R., Paterson, S. Validated Method for the Quantification of Buprenorphine in Postmortem Blood Using Solid-Phase Extraction and Two-Dimensional Gas Chromatography–Mass Spectrometry. J. Anal. Toxicol. (2015). 39: 519–525. |
| [179] | Ginsberg, G., Hattis, D., Russ, A., Sonawane, B., Physiologically Based Pharmacokinetic (PBPK) Modeling of Caffeine and Theophylline in Neonates and Adults: Implications for Assessing Children’s Risks from Environmental Agents. J. Toxicol. Environ. Health. Part A, (2004). 67: 297–329. |
| [180] | Edginton, A. N., Schmitt, W., Willmann, S., Development and Evaluation of a Generic Physiologically Based Pharmacokinetic Model for Children. Clin. Pharmacokinet. (2006). 45: 1013–1034. |
| [181] | Hebert, M. F., Easterling, T. R., Kirby, B., Carr, D. B., Buchanan, M. L., Rutherford, T., Thummel, K. E., Fishbein, D. P., Unadkat, J. D., Effects of pregnancy on CYP3A and P-glycoprotein activities as measured by disposition of midazolam and digoxin: a University of Washington specialized center of research study. Clin. Pharmacol. Ther. (2008). 84: 248–253. |
| [182] | Kanto, J., Sjövall, S., Erkkola, R., Himberg, J. J., Kangas, L., Placental transfer and maternal midazolam kinetics. Clin Pharmacol. Ther. (1983). 33: 786–791. |
| [183] | OECD. Guidance Document on the Characterisation, Validation and Reporting of Physiologically Based Kinetic (pbk) Models for Regulatory Purposes. OECD Series on Testing and Assessment, No. 331, OECD Series on Testing and Assessment 103. (2021). |
| [184] | Clewell III, H. J., Reddy, M. B., Lave, T., Andersen, M. E., Physiologically Based Pharmacokinetic Modeling, in: Gad, S. C. (Ed.), Preclinical Development Handbook: ADME and Biopharmaceutical Properties, Pharmaceutical Development Series. (2008). p. 1165 - 1225. |
| [185] | Coker, J. L., Ray-Griffith, S. L., McLeod, C., Han, X., Mancino, M., Kearns, G. L., Stowe, Z. N., Clearance of buprenorphine during pregnancy and neonatal outcomes. Arch. Womens. Ment. Health. (2021). 24: 933–939. |
| [186] | Honda, M., Omori, Y., Minei, S., Oshiyama, T., Shimizu, M., Sanaka, M., Kohama, T., Nakabayashi, M., Hirata, Y., Quantitative analysis of serum alpha 1-acid glycoprotein levels in normal and diabetic pregnancy. Diabetes Res. Clin. Pract (1990). 10: 147–152. |
| [187] | Bhatia, P., Chhabra, S., Physiological and anatomical changes of pregnancy: Implications for anaesthesia. Indian J. Anaesth. (2018). 62: 651–657. |
| [188] | Caritis, S. N., Sharma, S., Venkataramanan, R., Hankins, G. D., Miodovnik, M., Hebert, M. F., Umans, J. G., Benedetti, T., Mattison, D., Zajicek, A., Fischer, D., Jackson, A., Eunice Kennedy Shriver National Institute of Child Health and Human Development Obstetrical-Fetal Pharmacology Research Units Network, Pharmacology and placental transport of 17-hydroxyprogesterone caproate in singleton gestation. Am. J. Obstet. Gynecol. (2012). 207: 398. e1–8. |
| [189] | Srinivas, N. R., Syed, M., Applicability of a Single Time Point Strategy for the Prediction of Area Under the Concentration Curve of Linezolid in Patients: Superiority of Ctrough- over Cmax-Derived Linear Regression Models. Drugs R D (2016). 16: 69–79. |
| [190] | Johnson, R. E., Jones, H. E., Jasinski, D. R., Svikis, D. S., Haug, N. A., Jansson, L. M., Kissin, W. B., Alpan, G., Lantz, M. E., Cone, E. J., Wilkins, D. G., Golden, A. S., Huggins, G. R., Lester, B. M., Buprenorphine treatment of pregnant opioid--dependent women: maternal and neonatal outcomes. Drug Alcohol Depend. (2001). 63: 97–103. |
| [191] | American Psychiatric Association Diagnostic and Statistical Manual of Mental Disorders: DSM-5. 5th edn. Washington, D.C.: American Psychiatric Publishing. (2013). |
| [192] | Jana, S., Paliwal, J., Molecular mechanisms of cytochrome p450 induction: potential for drug-drug interactions. Curr. Protein Pept. Sci. (2007). 8: 619–628. |
| [193] | Centers for Disease Control and Prevention (CDC). Updated guidelines for the use of rifabutin or rifampin for the treatment and prevention of tuberculosis among HIV-infected patients taking protease inhibitors or nonnucleoside reverse transcriptase inhibitors. MMWR Morb Mortal Wkly Rep (2000). 49: 185–189. |
| [194] | Gallicano, K. D., Sahai, J., Shukla, V. K., Seguin, I., Pakuts, A., Kwok, D., Foster, B. C., Cameron, D. W., Induction of zidovudine glucuronidation and amination pathways by rifampicin in HIV-infected patients. Br J Clin. Pharmacol. (1999). 48: 168–179. |
| [195] | Oesch, F., Arand, M., Benedetti, M. S., Castelli, M. G., Dostert, P., Inducing properties of rifampicin and rifabutin for selected enzyme activities of the cytochrome P-450 and UDP-glucuronosyltransferase superfamilies in female rat liver. J. Antimicrob. Chemother. (1996). 37: 1111–1119. |
| [196] | Peters, S. A., Physiologically-Based Pharmacokinetic (pbpk) Modelling and Simulations. John Wiley & Sons, Inc., United States of America. (2012). p187 – 207. |
| [197] | Gordi, T., Xie, R., Huong, N. V., Huong, D. X., Karlsson, M. O., Ashton, M., A semiphysiological pharmacokinetic model for artemisinin in healthy subjects incorporating autoinduction of metabolism and saturable first-pass hepatic extraction. Br. J. Clin. Pharmacol. (2005). 59: 189–198. |
| [198] | Weinberg, D. S., Inturrisi, C. E., Reidenberg, B., Moulin, D. E., Nip, T. J., Wallenstein, S., Houde, R. W., Foley, K. M., Sublingual absorption of selected opioid analgesics. Clin. Pharmacol. Ther. (1988). 44: 335–342. |
| [199] | Niemi, M., Backman, J. T., Fromm, M. F., Neuvonen, P. J., Kivistö, K. T., Pharmacokinetic Interactions with Rifampicin. Clin. Pharmacokinet. (2003). 42: 819–850. |
| [200] | McCance-Katz, E. F., Moody, D. E., Prathikanti, S., Friedland, G., Rainey, P. M., Rifampin, but not rifabutin, may produce opiate withdrawal in buprenorphine-maintained patients. Drug Alcohol Depend. (2011). 118: 326–334. |
| [201] | Hagelberg, N. M., Fihlman, M., Hemmilä, T., Backman, J. T., Laitila, J., Neuvonen, P. J., Laine, K., Olkkola, K. T., Saari, T. I., Rifampicin decreases exposure to sublingual buprenorphine in healthy subjects. Pharmacol. Res. Perspect. (2016). e00271. |
| [202] | Zhou, S., Yung Chan, S., Cher Goh, B., Chan, E., Duan, W., Huang, M., McLeod, H. L. Mechanism-based inhibition of cytochrome P450 3A4 by therapeutic drugs. Clin. Pharmacokinet. (2005). 44: 279–304. |
| [203] | Rowland Yeo, K., Walsky, R. L., Jamei, M., Rostami-Hodjegan, A., Tucker, G. T., Prediction of time-dependent CYP3A4 drug-drug interactions by physiologically based pharmacokinetic modelling: impact of inactivation parameters and enzyme turnover. Eur. J. Pharm. Sci. (2011). 43: 160–173. |
| [204] | Paine, M. F., Hart, H. L., Ludington, S. S., Haining, R. L., Rettie, A. E., Zeldin, D. C., The human intestinal cytochrome P450 “pie.” Drug Metab. Dispos. (2006). 34: 880–886. |
| [205] | Zanger, U. M., Schwab, M., Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. (2013). 138: 103–141. |
| [206] | Owens, R. C., Nolin, T. D., Antimicrobial-associated QT interval prolongation: pointes of interest. Clin. Infect. Dis. (2006). 43: 1603–1611. |
| [207] | Dresser, G. K., Spence, J. D., Bailey, D. G. Pharmacokinetic-Pharmacodynamic Consequences and Clinical Relevance of Cytochrome P450 3A4 Inhibition. Clin. Pharmacokinet. (2000). 38: 41–57. |
| [208] | Owens, R. C. QT prolongation with antimicrobial agents: understanding the significance. Drugs (2004). 64: 1091–1124. |
| [209] | van Haarst, A. D., van ’t Klooster, G. A., van Gerven, J. M., Schoemaker, R. C., van Oene, J. C., Burggraaf, J., Coene, M. C., Cohen, A. F., The influence of cisapride and clarithromycin on QT intervals in healthy volunteers. Clin. Pharmacol. Ther. (1998). 64: 542–546. |
| [210] | Simmat-Durand, L., Lejeune, C., Gourarier, L., Pregnancy under high-dose buprenorphine. Eur. J. Obstet. Gynecol. Reprod. Biol. (2009). 142: 119–123. |
| [211] | O’Connor, A. B., O’Brien, L., Alto, W. A., Maternal Buprenorphine Dose at Delivery and Its Relationship to Neonatal Outcomes. EAR (2016). 22: 127–130. |
| [212] | Jansson, L. M., Velez, M. L., McConnell, K., Milio, L., Spencer, N., Jones, H., DiPietro, J. A., Maternal buprenorphine treatment during pregnancy and maternal physiology. Drug Alcohol Depend. (2019). 201: 38–44. |
| [213] | Cone, E. J., Gorodetzky, C. W., Yousefnejad, D., Buchwald, W. F., Johnson, R. E., The metabolism and excretion of buprenorphine in humans. Drug. Metab. Dispos. (1984). 12: 577–581. |
| [214] | García-Martín, E., Martínez, C., Ladero, J. M., Agúndez, J. A. G., Interethnic and intraethnic variability of CYP2C8 and CYP2C9 polymorphisms in healthy individuals. Mol. Diagn. Ther. (2006). 10: 29–40. |
| [215] | Goldstein, J. A., de Morais, S. M., Biochemistry and molecular biology of the human CYP2C subfamily. Pharmacogenetics (1994). 4: 285–299. |
| [216] | Rowland, A., Miners, J. O., Mackenzie, P. I., The UDP-glucuronosyltransferases: their role in drug metabolism and detoxification. Int. J. Biochem. Cell Biol. (2013). 45: 1121–1132. |
| [217] | Savage, S. R., Long-term opioid therapy: assessment of consequences and risks. J. Pain Symptom Manage. (1996). 11: 274– 286. |
| [218] | Tzschentke, T. M., Behavioral pharmacology of buprenorphine, with a focus on preclinical models of reward and addiction. Psychopharmacology (Berl) (2002). 161: 1–16. |
| [219] | Robinson, S. E., Wallace, M. J., Effect of perinatal buprenorphine exposure on development in the rat. J. Pharmacol. Exp. Ther. (2001). 298: 797–804. |
| [220] | Kintz, P., A New Series of 13 Buprenorphine-Related Deaths. Clin. Biochem. (2002). 35: 513–516. |
| [221] | Kintz, P., Deaths involving buprenorphine: a compendium of French cases. Excerpts from TIAFT 2000. Forensic Sci.Int. (2001). 121: 65–69. |
| [222] | Pelissier-Alicot, A.-L., Sastre, C., Baillif-Couniou, V., Gaulier, J.-M., Kintz, P., Kuhlmann, E., Perich, P., Bartoli, C., Piercecchi-Marti, M.-D., Leonetti, G., Buprenorphine-related deaths: unusual forensic situations. Int. J. Legal Med. (2010). 124: 647–651. |
| [223] | Tracqui, A., Kintz, P., Ludes, B., Buprenorphine-Related Deaths Among Drug Addicts in France: A Report on 20 Fatalities. J. Anal. Toxicol. (1998). 22: 430–434. |
| [224] | Ross, D., High dose buprenorphine in pregnancy. ANZJOG (2004). 44: 80–80. |
| [225] | Kleber, H. D., Pharmacologic treatments for opioid dependence: detoxification and maintenance options. Dialogues Clin. Neurosci. (2007). 9: 455–470. |
| [226] | Sigmon, S. C., Bisaga, A., Nunes, E. V., O’Connor, P. G., Kosten, T., Woody, G., Opioid detoxification and naltrexone induction strategies: recommendations for clinical practice. Am. J. Drug Alcohol Abuse (2012). 38: 187–199. |
| [227] | Badhan, R. K. S., Gittins, R., Al Zabit, D., The optimization of methadone dosing whilst treating with rifampicin: A pharmacokinetic modeling study. Drug. Alcohol. Depend. (2019). 200: 168–180. |
| [228] | Bogen, D. L., Perel, J. M., Helsel, J. C., Hanusa, B. H., Romkes, M., Nukui, T., Friedman, C. R., Wisner, K. L., Pharmacologic evidence to support clinical decision making for peripartum methadone treatment. Psychopharmacology (Berl.) (2013). 225: 441–451. |
| [229] | Jones, H. E., Johnson, R. E., O’Grady, K. E., Jasinski, D. R., Tuten, M., Milio, L., Dosing adjustments in postpartum patients maintained on buprenorphine or methadone. J. Addict. Med. (2008). 2: 103–107. |
| [230] | Pace, C. A., Kaminetzky, L. B., Winter, M., Cheng, D. M., Saia, K., Samet, J. H., Walley, A. Y. Postpartum changes in methadone maintenance dose. J. Subst. Abuse Treat. (2014). 47: 229–232. |
| [231] | Pan, X., Yamazaki, S., Neuhoff, S., Zhang, M., Pilla Reddy, V., Unraveling pleiotropic effects of rifampicin by using physiologically based pharmacokinetic modeling: Assessing the induction magnitude of P-glycoprotein–cytochrome P450 3A4 dual substrates. CPT Pharmacometrics Syst. Pharmacol. (2021). 10: 1485–1496. |
| [232] | Brown, S. M., Campbell, S. D., Crafford, A., Regina, K. J., Holtzman, M. J., Kharasch, E. D., P-Glycoprotein Is a Major Determinant of Norbuprenorphine Brain Exposure and Antinociception. J. Pharmacol. Exp. Ther. (2012). 343: 53–61. |
| [233] | Chang, Y., Moody, D. E., McCance-Katz, E. F., Novel Metabolites of Buprenorphine Detected in Human Liver Microsomes and Human Urine. Drug. Metab. Dispos. (2006). 34: 440–448. |
| [234] | Liao, M. Z., Gao, C., Shireman, L. M., Phillips, B., Risler, L. J., Neradugomma, N. K., Choudhari, P., Prasad, B., Shen, D. D., Mao, Q., P-gp/ABCBl Exerts Differential Impacts On Brain and Fetal Exposure to Norbuprenorphine. Pharmacol. Res. (2017). 119: 61–71. |
| [235] | Darwich, A. S., Aslam, U., Ashcroft, D. M., Rostami-Hodjegan, A., Meta-Analysis of the Turnover of Intestinal Epithelia in Preclinical Animal Species and Humans. Drug Metab. Dispos. (2014). 42: 2016–2022. |
| [236] | Lindemalm, S., Nydert, P., Svensson, J.-O., Stahle, L., Sarman, I., Transfer of Buprenorphine Into Breast Milk and Calculation of Infant Drug Dose. J. Hum. Lact. (2009). 25: 199–205. |
| [237] | Ilett, K. F., Hackett, L. P., Gower, S., Doherty, D. A., Hamilton, D., Bartu, A. E., Estimated dose exposure of the neonate to buprenorphine and its metabolite norbuprenorphine via breastmilk during maternal buprenorphine substitution treatment. Breastfeed Med. (2012). 7: 269–274. |
| [238] | Tracy, T. S., Venkataramanan, R., Glover, D. D., Caritis, S. N., National Institute for Child Health and Human Development Network of Maternal-Fetal-Medicine Units, Temporal changes in drug metabolism (CYP1A2, CYP2D6 and CYP3A Activity) during pregnancy. Am. J. Obstet. Gynecol. (2005). 192: 633–639. |
APA Style
Nnanna, T. B. (2024). Clinical Therapy Dose Optimization of Sublingual Buprenorphine in Poorly Adherent Pregnant Patients: A PBPK Translational Modelling Study. International Journal of Pharmacy and Chemistry, 10(4), 46-79. https://doi.org/10.11648/j.ijpc.20241004.11
ACS Style
Nnanna, T. B. Clinical Therapy Dose Optimization of Sublingual Buprenorphine in Poorly Adherent Pregnant Patients: A PBPK Translational Modelling Study. Int. J. Pharm. Chem. 2024, 10(4), 46-79. doi: 10.11648/j.ijpc.20241004.11
AMA Style
Nnanna TB. Clinical Therapy Dose Optimization of Sublingual Buprenorphine in Poorly Adherent Pregnant Patients: A PBPK Translational Modelling Study. Int J Pharm Chem. 2024;10(4):46-79. doi: 10.11648/j.ijpc.20241004.11
@article{10.11648/j.ijpc.20241004.11,
author = {Tobechi Brendan Nnanna},
title = {Clinical Therapy Dose Optimization of Sublingual Buprenorphine in Poorly Adherent Pregnant Patients: A PBPK Translational Modelling Study
},
journal = {International Journal of Pharmacy and Chemistry},
volume = {10},
number = {4},
pages = {46-79},
doi = {10.11648/j.ijpc.20241004.11},
url = {https://doi.org/10.11648/j.ijpc.20241004.11},
eprint = {https://article.sciencepublishinggroup.com/pdf/10.11648.j.ijpc.20241004.11},
abstract = {Plasma levels of sublingual buprenorphine utilized in the therapy of opioid use disorder, has been demonstrated to undergo gestation-associated decline in vivo, to an extent influenced by upheavals physiologically across gestational trimesters. However, based on extant literature, a dearth of knowledge exists in the optimization of buprenorphine therapeutic modalities, pharmacokinetic interactions and posological scrutiny, necessary for successful regimen adherence. A physiologically-based pharmacokinetic modelling methodology in a virtual clinical trial premise was utilized to investigate gestational upheavals in peak plasma buprenorphine concentrations, followed by a pharmacokinetic drug-drug interaction investigation and dose optimization strategy, to maintain buprenorphine levels above proposed thresholds of 1ng/ml and below 22.2ng/ml adjudicated as a fatality limit. A fold decline (> 1.3fold) in buprenorphine mean peak plasma concentration (92% - 74%) was evident for the model predicted buprenorphine metrics across selected gestational weeks to term in line with the model predicted increases in physiological upheavals occurring across gestation which may influence the changes. The rifampicin mediated drug-drug interaction on buprenorphine levels initially resulted in fold decreases (>1.5 fold) over a twenty-four hour duration, in concert with escalating physiological metrics across gestational trimesters. The interaction perpetrated with Clarithromycin dosing resulted in fold increases (> 2-fold) in the plasma concentration as well as an increase in other metrics associated with buprenorphine kinetics. The dose optimization approach maintained majority of subjects (>90%) with the extensive metabolizer (EM) phenotype above 1ng/ml and below 22.2ng/ml in the 8mg – 24mg dose ranges albeit with 1% and 3% in the 28mg and 32mg doses above the fatality limit respectively. This study demonstrates the utility of physiologically based pharmacokinetic methods to predict the time course of administered buprenorphine in plasma during gestation which could aid clinician decisions in a translational manner, in order to optimize therapeutic modalities in the therapy of opioid use disorder.
},
year = {2024}
}
TY - JOUR T1 - Clinical Therapy Dose Optimization of Sublingual Buprenorphine in Poorly Adherent Pregnant Patients: A PBPK Translational Modelling Study AU - Tobechi Brendan Nnanna Y1 - 2024/12/31 PY - 2024 N1 - https://doi.org/10.11648/j.ijpc.20241004.11 DO - 10.11648/j.ijpc.20241004.11 T2 - International Journal of Pharmacy and Chemistry JF - International Journal of Pharmacy and Chemistry JO - International Journal of Pharmacy and Chemistry SP - 46 EP - 79 PB - Science Publishing Group SN - 2575-5749 UR - https://doi.org/10.11648/j.ijpc.20241004.11 AB - Plasma levels of sublingual buprenorphine utilized in the therapy of opioid use disorder, has been demonstrated to undergo gestation-associated decline in vivo, to an extent influenced by upheavals physiologically across gestational trimesters. However, based on extant literature, a dearth of knowledge exists in the optimization of buprenorphine therapeutic modalities, pharmacokinetic interactions and posological scrutiny, necessary for successful regimen adherence. A physiologically-based pharmacokinetic modelling methodology in a virtual clinical trial premise was utilized to investigate gestational upheavals in peak plasma buprenorphine concentrations, followed by a pharmacokinetic drug-drug interaction investigation and dose optimization strategy, to maintain buprenorphine levels above proposed thresholds of 1ng/ml and below 22.2ng/ml adjudicated as a fatality limit. A fold decline (> 1.3fold) in buprenorphine mean peak plasma concentration (92% - 74%) was evident for the model predicted buprenorphine metrics across selected gestational weeks to term in line with the model predicted increases in physiological upheavals occurring across gestation which may influence the changes. The rifampicin mediated drug-drug interaction on buprenorphine levels initially resulted in fold decreases (>1.5 fold) over a twenty-four hour duration, in concert with escalating physiological metrics across gestational trimesters. The interaction perpetrated with Clarithromycin dosing resulted in fold increases (> 2-fold) in the plasma concentration as well as an increase in other metrics associated with buprenorphine kinetics. The dose optimization approach maintained majority of subjects (>90%) with the extensive metabolizer (EM) phenotype above 1ng/ml and below 22.2ng/ml in the 8mg – 24mg dose ranges albeit with 1% and 3% in the 28mg and 32mg doses above the fatality limit respectively. This study demonstrates the utility of physiologically based pharmacokinetic methods to predict the time course of administered buprenorphine in plasma during gestation which could aid clinician decisions in a translational manner, in order to optimize therapeutic modalities in the therapy of opioid use disorder. VL - 10 IS - 4 ER -
Clinical Pharmacology & Pharmaceutical Research Unit, PYCAD Pharmacy, Nigeria
Biography: Tobechi Brendan Nnanna is a pharmacist professional with over two years of expertise in pharmacokinetic (PK) and pharmacodynamic (PD) modelling, PBPK simulation, and statistical data analysis. Holding a Master of Science in Pharmacokinetics (Distinction) from Aston University, United Kingdom, and a Bachelor of Pharmacy from Madonna University, Nigeria, he demonstrates a strong academic foundation enhanced by the prestigious Ferguson Scholarship Award. Professionally, Tobechi is currently engaged as a Clinical Pharmacology Researcher(freelance), leveraging advanced PK modelling tools such as SimCYP, PK-Sim, Mobi, nlMixR for drug distribution studies and clinical outcome predictions as well as other novel published open-source state-of-the art software. His work emphasizes translational modelling, population PK, clinical pharmacology to improve therapeutic efficacy. Previously, he contributed to pharmaceutical projects as a Consultant Pharmacist, guiding clinical insights and mentoring junior professionals. With a passion for data-driven & lab-based research, Tobechi strives to optimize therapeutic strategies through innovative pharmacometrics approaches and collaborative multidisciplinary efforts.
Research Fields: Pharmacokinetics in Special Population, Drug-Drug Interaction Mechanism, Personalized Medicine & Pharmacogenomics, Toxicokinetics & Safety Evaluation Studies, Drug Metabolism & Enzyme Kinetics, Population Pharmacokinetics, Pharmacometrics, Preclinical Pharmacokinetic & Pharmacodynamic Correlation, Physiologically based Pharmacokinetic Modeling, Pharmacokinetic Modelling & Simulation


Figure 1. A model workflow for buprenorphine.
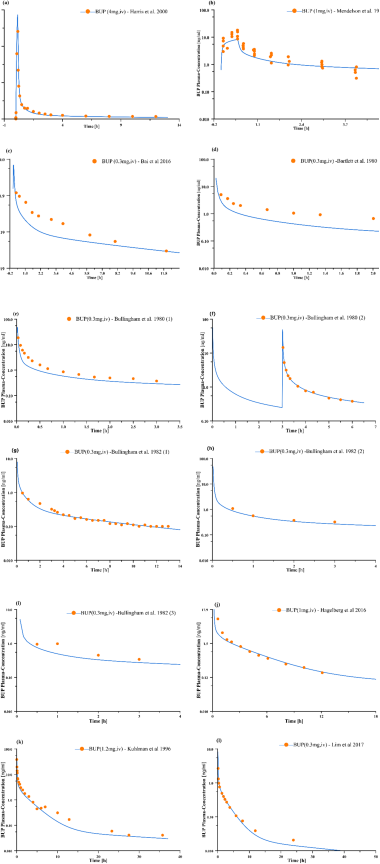
Figure 2. Simulated buprenorphine plasma concentrations compared to clinical plasma concentrations derived from retrospective studies.
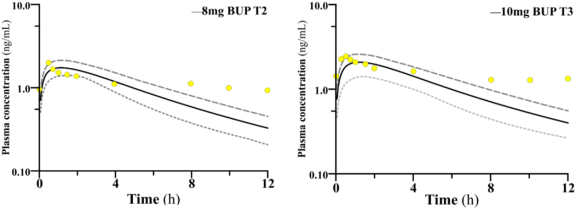
Figure 3. Predicted Log of buprenorphine plasma concentration-time profiles in gestational trimesters. Second (T2) and third (T3) compared to clinical plasma concentrations derived from a retrospective study [173]. Solid black line depicts logged mean predicted concentration-time profile. Dotted lines (orange and ash) represent the 95th and 5th percentiles; T: Trimester.
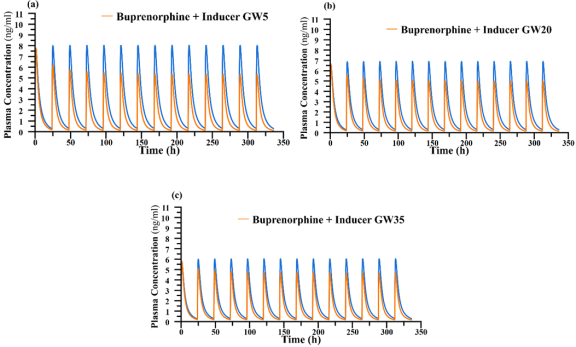
Figure 4. (A - C). Simulated mean concentration-time profiles of buprenorphine daily doses (16mg) in presence and absence of Rifampicin (600). GW – gestational week; b.d – twice daily dosing; Orange line displayed as ‘DDI’; Blue Line depicted as ‘No DDI’.
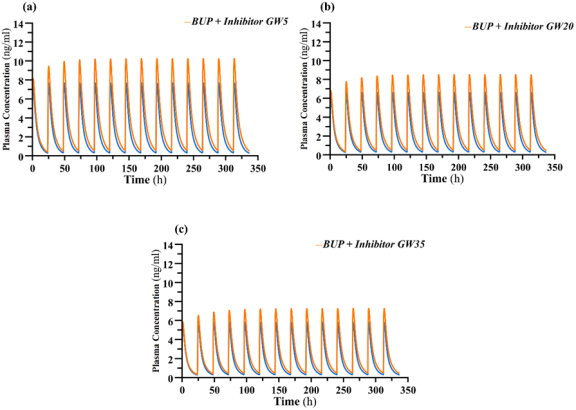
Figure 5. (A - C). Simulated mean concentration-time profiles of buprenorphine daily doses (16mg) in presence and absence of Clarithromycin. GW – gestational week; b.d – twice daily dosing; Orange line displayed as ‘DDI’; Blue Line depicted as ‘No DDI’.
Information

