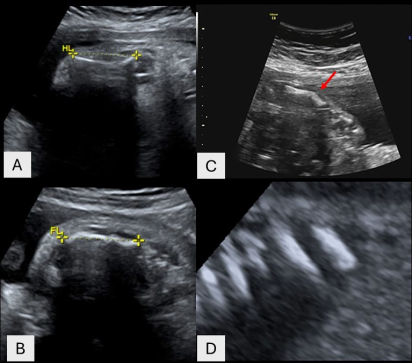
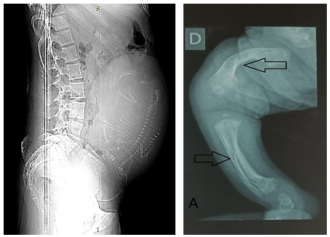
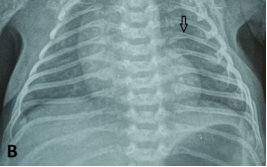
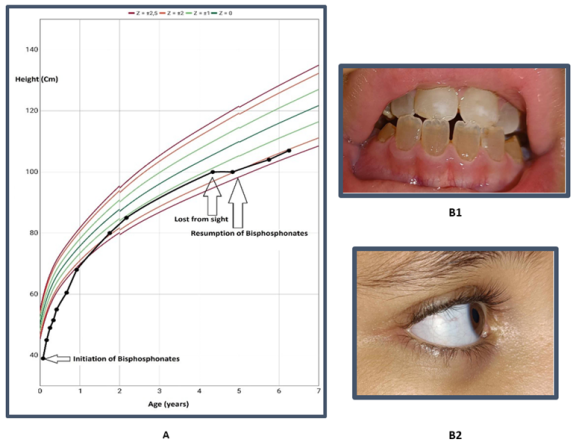
Osteogenesis imperfecta (OI) is a genetically heterogeneous hereditary disorder characterized by increased bone fragility, leading to recurrent fractures following minimal or no trauma. In 2009, the International Nomenclature Committee for Constitutional Disorders of the Skeleton (INCDS), and as published in the 2010 Nosology, OI was reclassified into five major types. Type IV OI is caused by mutations in the COL1A1 and COL1A2 genes, which are responsible for the synthesis of type I collagen, a critical component of bone matrix. These mutations result in varying degrees of bone deformity, growth retardation, and other systemic manifestations. In this report, we present a case of antenatal diagnosis of OI, providing a comprehensive overview of the disease's evolution from the intrauterine stage through early childhood and into school age. This case illustrates the wide phenotypic variability of OI, with clinical manifestations spanning the prenatal period, birth, infancy, and school years, and highlights the multiple complications encountered at each stage of development. The profound impact of the disorder on the patient’s daily functioning and the family’s quality of life is also discussed.
| Published in | American Journal of Pediatrics (Volume 11, Issue 3) |
| DOI | 10.11648/j.ajp.20251103.19 |
| Page(s) | 173-178 |
| Creative Commons |
This is an Open Access article, distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution and reproduction in any medium or format, provided the original work is properly cited. |
| Copyright |
Copyright © The Author(s), 2025. Published by Science Publishing Group |
Children, Osteogenesis Imperfecta, Growth Retardation, Pathological Fractures, Antenatal Diagnosis
OI | Osteogenesis imperfecta |
CT | Computed Tomography |
SD | Standard Deviation |
| [1] | Subramanian S, Anastasopoulou C, Viswanathan VK. Osteogenesis Imperfecta. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 [cité 3 juill 2025]. Disponible sur: |
| [2] | Marini JC, Dang Do AN. Osteogenesis Imperfecta. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, et al., éditeurs. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000 [cité 23 mai 2024]. Disponible sur: |
| [3] | Deguchi M, Tsuji S, Katsura D, Kasahara K, Kimura F, Murakami T. Current Overview of Osteogenesis Imperfecta. Medicina (Mex). 10 mai 2021; 57(5): 464. |
| [4] | Rodriguez Celin M, Steiner RD, Basel D. COL1A1- and COL1A2-Related Osteogenesis Imperfecta. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, éditeurs. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993 [cité 3 juill 2025]. Disponible sur: |
| [5] | Van Dijk F s., Sillence D o. Osteogenesis imperfecta: Clinical diagnosis, nomenclature and severity assessment. Am J Med Genet A. 2014; 164(6): 1470-81. |
| [6] | Costache A, Riza AL, Popescu M, Șerban RC, Mituț-Velișcu AM, Streață I. Diagnostic Challenges in Bone Fragility: Osteogenesis Imperfecta Case Series. Biomedicines. avr 2025; 13(4): 865. |
| [7] | Marini JC, Forlino A, Bächinger HP, Bishop NJ, Byers PH, Paepe AD, et al. Osteogenesis imperfecta. Nat Rev Dis Primer. 18 août 2017; 3: 17052. |
| [8] | Van Dijk F, Sillence D. Osteogenesis imperfecta: Clinical diagnosis, nomenclature and severity assessment. Am J Med Genet A. juin 2014; 164(6): 1470-81. |
| [9] | Rauch F, Glorieux FH. Osteogenesis imperfecta. Lancet Lond Engl. 24 avr 2004; 363(9418): 1377-85. |
| [10] | Biggin A, Munns CF. Osteogenesis imperfecta: diagnosis and treatment. Curr Osteoporos Rep. sept 2014; 12(3): 279-88. |
| [11] | Lim J, Grafe I, Alexander S, Lee B. Genetic Causes and Mechanisms of Osteogenesis Imperfecta. Bone. sept 2017; 102: 40-9. |
| [12] | Panzaru MC, Florea A, Caba L, Gorduza EV. Classification of osteogenesis imperfecta: Importance for prophylaxis and genetic counseling. World J Clin Cases. 26 avr 2023; 11(12): 2604-20. |
| [13] |
Kahyaoğlu S, Danışman N. Clinical and ultrasonographic hints for prenatal diagnosis and management of the lethal skeletal dysplasias: a review of the current literature. Acta Medica [Internet]. 9 oct 2012 [cité 18 juill 2025]; 43(1). Disponible sur:
https://www.actamedica.org/index.php/actamedica/article/view/165 |
| [14] | Marom R, Rabenhorst BM, Morello R. Osteogenesis imperfecta: an update on clinical features and therapies. Eur J Endocrinol. oct 2020; 183(4): R95-106. |
| [15] | Zaripova AR, Khusainova RI. Modern classification and molecular-genetic aspects of osteogenesis imperfecta. Vavilovskii Zhurnal Genet Sel. mars 2020; 24(2): 219-27. |
| [16] | Bellur S, Jain M, Cuthbertson D, Krakow D, Shapiro JR, Steiner RD, et al. Cesarean delivery is not associated with decreased at-birth fracture rates in osteogenesis imperfecta. Genet Med Off J Am Coll Med Genet. juin 2016; 18(6): 570-6. |
| [17] | Rossi V, Lee B, Marom R. Osteogenesis imperfecta: advancements in genetics and treatment. Curr Opin Pediatr. déc 2019; 31(6): 708-15. |
| [18] | Chen CP, Lin SP, Su YN, Chern SR, Lin MH, Su JW, et al. Osteogenesis imperfecta type IV: prenatal molecular diagnosis and genetic counseling in a pregnancy carried to full term with favorable outcome. Taiwan J Obstet Gynecol. juin 2012; 51(2): 271-5. |
| [19] | Cubert R, Cheng EY, Mack S, Pepin MG, Byers PH. Osteogenesis imperfecta: mode of delivery and neonatal outcome. Obstet Gynecol. janv 2001; 97(1): 66-9. |
| [20] | Wang H, Huang X, Wu A, Li Q. Management of anesthesia in a patient with osteogenesis imperfecta and multiple fractures: a case report and review of the literature. J Int Med Res. juin 2021; 49(6): 3000605211028420. |
| [21] | Hidalgo Perea S, Green DW. Osteogenesis imperfecta: treatment and surgical management. Curr Opin Pediatr. 1 févr 2021; 33(1): 74-8. |
| [22] | Majdoub F, Ferjani HL, Nessib DB, Kaffel D, Maatallah K, Hamdi W. Denosumab use in osteogenesis imperfecta: an update on therapeutic approaches. Ann Pediatr Endocrinol Metab. juin 2023; 28(2): 98-106. |
| [23] | Forlino A, Marini JC. Osteogenesis imperfecta. Lancet Lond Engl. 16 avr 2016; 387(10028): 1657-71. |
| [24] | Jain M, Tam A, Shapiro JR, Steiner RD, Smith PA, Bober MB, et al. Growth characteristics in individuals with osteogenesis imperfecta in North America: results from a multicenter study. Genet Med Off J Am Coll Med Genet. févr 2019; 21(2): 275-83. |
| [25] | Caudevilla Lafuente P, de Arriba Muñoz A, Izquierdo Álvarez S, Ferrer Lozano M, Medrano San Ildefonso M, Labarta Aizpún JI. Osteogenesis imperfecta: Review of 40 patients. Med Clin (Barc). 1 juin 2020; 154(12): 512-8. |
| [26] | Botor M, Fus-Kujawa A, Uroczynska M, Stepien KL, Galicka A, Gawron K, et al. Osteogenesis Imperfecta: Current and Prospective Therapies. Biomolecules. 10 oct 2021; 11(10): 1493. |
| [27] | Hill CL, Baird WO, Walters SJ. Quality of life in children and adolescents with Osteogenesis Imperfecta: a qualitative interview based study. Health Qual Life Outcomes. 16 avr 2014; 12: 54. |
| [28] | Arundel P, Borg SA. Early Life Management of Osteogenesis Imperfecta. Curr Osteoporos Rep. 2023; 21(6): 779-86. |
APA Style
Jelalia, N., Marzouk, A., Thebti, R., Jallouli, L., Friha, F., et al. (2025). Diagnosis and Management of Type 4-IV Osteogenesis Imperfecta from Intrauterine Life to School Age: A Clinical Case Study. American Journal of Pediatrics, 11(3), 173-178. https://doi.org/10.11648/j.ajp.20251103.19
ACS Style
Jelalia, N.; Marzouk, A.; Thebti, R.; Jallouli, L.; Friha, F., et al. Diagnosis and Management of Type 4-IV Osteogenesis Imperfecta from Intrauterine Life to School Age: A Clinical Case Study. Am. J. Pediatr. 2025, 11(3), 173-178. doi: 10.11648/j.ajp.20251103.19
AMA Style
Jelalia N, Marzouk A, Thebti R, Jallouli L, Friha F, et al. Diagnosis and Management of Type 4-IV Osteogenesis Imperfecta from Intrauterine Life to School Age: A Clinical Case Study. Am J Pediatr. 2025;11(3):173-178. doi: 10.11648/j.ajp.20251103.19
@article{10.11648/j.ajp.20251103.19,
author = {Nour Jelalia and Asma Marzouk and Rahma Thebti and Leila Jallouli and Farida Friha and Ahlem Kefi and Asma Bouaziz},
title = {Diagnosis and Management of Type 4-IV Osteogenesis Imperfecta from Intrauterine Life to School Age: A Clinical Case Study
},
journal = {American Journal of Pediatrics},
volume = {11},
number = {3},
pages = {173-178},
doi = {10.11648/j.ajp.20251103.19},
url = {https://doi.org/10.11648/j.ajp.20251103.19},
eprint = {https://article.sciencepublishinggroup.com/pdf/10.11648.j.ajp.20251103.19},
abstract = {Osteogenesis imperfecta (OI) is a genetically heterogeneous hereditary disorder characterized by increased bone fragility, leading to recurrent fractures following minimal or no trauma. In 2009, the International Nomenclature Committee for Constitutional Disorders of the Skeleton (INCDS), and as published in the 2010 Nosology, OI was reclassified into five major types. Type IV OI is caused by mutations in the COL1A1 and COL1A2 genes, which are responsible for the synthesis of type I collagen, a critical component of bone matrix. These mutations result in varying degrees of bone deformity, growth retardation, and other systemic manifestations. In this report, we present a case of antenatal diagnosis of OI, providing a comprehensive overview of the disease's evolution from the intrauterine stage through early childhood and into school age. This case illustrates the wide phenotypic variability of OI, with clinical manifestations spanning the prenatal period, birth, infancy, and school years, and highlights the multiple complications encountered at each stage of development. The profound impact of the disorder on the patient’s daily functioning and the family’s quality of life is also discussed.},
year = {2025}
}
TY - JOUR T1 - Diagnosis and Management of Type 4-IV Osteogenesis Imperfecta from Intrauterine Life to School Age: A Clinical Case Study AU - Nour Jelalia AU - Asma Marzouk AU - Rahma Thebti AU - Leila Jallouli AU - Farida Friha AU - Ahlem Kefi AU - Asma Bouaziz Y1 - 2025/08/05 PY - 2025 N1 - https://doi.org/10.11648/j.ajp.20251103.19 DO - 10.11648/j.ajp.20251103.19 T2 - American Journal of Pediatrics JF - American Journal of Pediatrics JO - American Journal of Pediatrics SP - 173 EP - 178 PB - Science Publishing Group SN - 2472-0909 UR - https://doi.org/10.11648/j.ajp.20251103.19 AB - Osteogenesis imperfecta (OI) is a genetically heterogeneous hereditary disorder characterized by increased bone fragility, leading to recurrent fractures following minimal or no trauma. In 2009, the International Nomenclature Committee for Constitutional Disorders of the Skeleton (INCDS), and as published in the 2010 Nosology, OI was reclassified into five major types. Type IV OI is caused by mutations in the COL1A1 and COL1A2 genes, which are responsible for the synthesis of type I collagen, a critical component of bone matrix. These mutations result in varying degrees of bone deformity, growth retardation, and other systemic manifestations. In this report, we present a case of antenatal diagnosis of OI, providing a comprehensive overview of the disease's evolution from the intrauterine stage through early childhood and into school age. This case illustrates the wide phenotypic variability of OI, with clinical manifestations spanning the prenatal period, birth, infancy, and school years, and highlights the multiple complications encountered at each stage of development. The profound impact of the disorder on the patient’s daily functioning and the family’s quality of life is also discussed. VL - 11 IS - 3 ER -
Pediatrics and Neonatology Departement, Faculty of Medecine of Tunis, Ben Arous, Tunisia
Pediatrics and Neonatology Departement, Faculty of Medecine of Tunis, Ben Arous, Tunisia
Pediatrics and Neonatology Departement, Faculty of Medecine of Tunis, Ben Arous, Tunisia
Information