Abstract
Anaplastic lymphoma kinase (ALK) is a clinically important therapeutic target in ALK-positive cancers, particularly non-small cell lung cancer (NSCLC). Although ALK tyrosine kinase inhibitors (ALK-TKIs) initially produce substantial clinical benefit, long-term efficacy is frequently compromised by the emergence of resistance mutations within the kinase domain. Among these, the solvent-front mutation G1202R and the gatekeeper mutation L1196M, especially when present as a compound mutation, pose significant challenges by markedly reducing inhibitor binding and therapeutic response. The present study aims to systematically investigate the structural and functional consequences of ALK G1202R and G1202R/L1196M resistance mutations using an integrated computational structural biology and CRISPR-Cas9 genome-editing framework. High-resolution crystal structures of mutant ALK in complex with the next-generation inhibitor NVL-655 (PDB ID: 9GBE) were analysed using PyMOL-based visualisation, B-factor analysis to assess conformational flexibility, MMDB annotations, and COSMIC-3D mutation mapping to identify mutation hotspots near ligand-binding regions. In parallel, allele-specific CRISPR-Cas9 single-guide RNAs were designed using E-CRISP and CHOPCHOP to enable precise modeling of these resistance mutations with minimal off-target effects. Structural analyses revealed that the G1202R mutation induces substantial steric hindrance and electrostatic alterations at the ATP-binding pocket, while the L1196M mutation stabilizes an active kinase conformation. Together, these changes destabilize inhibitor interactions and significantly impair binding of earlier-generation ALK-TKIs. In contrast, next-generation inhibitors such as NVL-655 and TPX-0131 demonstrate improved accommodation of the altered binding pocket, consistent with emerging preclinical and clinical observations. Overall, this study provides mechanistic insight into ALK resistance evolution by integrating structural modeling with genome-editing strategies. The proposed framework provides a robust platform for studying compound resistance mutations and supports the rational design and validation of next-generation ALK inhibitors and CRISPR-based therapeutic strategies for ALK-driven cancers that are resistant to these agents.
|
Published in
|
American Journal of BioScience (Volume 14, Issue 1)
|
|
DOI
|
10.11648/j.ajbio.20261401.12
|
|
Page(s)
|
8-19 |
|
Creative Commons
|

This is an Open Access article, distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution and reproduction in any medium or format, provided the original work is properly cited.
|
|
Copyright
|
Copyright © The Author(s), 2026. Published by Science Publishing Group
|
Keywords
ALK Kinase Domain, G1202R/L1196M Mutations, NVL-655 Inhibitor, B-factor Analysis, CRISPR-Cas9 Genome Editing, Structural Bioinformatics, Drug Resistance Mechanisms
1. Introduction
Anaplastic lymphoma kinase (ALK) rearrangement defines an evident molecular form of non–small cell lung cancer (NSCLC) that responds initially and often dramatically to ALK tyrosine kinase inhibitors (ALK-TKIs). However, the long-term efficacy of ALK-targeted therapy is limited by the emergence of secondary resistance mutations within the ALK kinase domain, particularly under TKI-selective pressure. Among these alterations, mutations affecting the solvent-front residue G
R and the gatekeeper residue L
M are consistently identified as two of the most clinically significant mediators of resistance to second- and third-generation inhibitors
| [1] | Awad, M. M., & Plotkin, S. (2023). Resistance mechanisms to next-generation ALK inhibitors in NSCLC. Nature Reviews Clinical Oncology, 20(3), 175–189.
https://doi.org/10.1038/s41571-022-00733-9 |
| [2] | Lin, J. J., Zhu, V. W., Marcoux, N., et al. (2023). Patterns of lorlatinib resistance mutations in ALK-positive lung cancer. Journal of Clinical Oncology, 41(12), 2190–2202.
https://doi.org/10.1200/JCO.22.01335 |
| [3] | Gainor, J. F., et al. (2024). Clinical landscape of ALK resistance mutations following sequential ALK-TKI therapy. Clinical Cancer Research, 30(4), 812–823.
https://doi.org/10.1158/1078-0432.CCR-23-0147 |
[1-3]
. When these mutations co-occur as a compound genotype (G
R/L
M), they confer a highly refractory phenotype that resists even advanced macrocyclic TKIs such as lorlatinib.
Recent clinical sequencing and liquid biopsy analyses (
) have confirmed that solvent-front mutation, especially G
R continue to dominate in patients progressing on lorlatinib and other next-generation inhibitors
. Structural and biochemical interpretation show that the massive, positively charged arginine introduced at position
disrupts the solvent-front architecture, altering both steric accessibility and local electrostatics, while the L
M substitution alters the hydrophobic gatekeeper environment and the geometry of the ATP-binding cleft.
The compound mutation synergistically perturbs the active-site conformation, producing a drastically remodelled pocket that reduces binding affinities across multiple small-molecule scaffolds
. This structural reconfiguration the importance of detailed visualization to understand how compound mutations distort the binding pocket and compromise inhibitor efficacy
.The availability of high-resolution structural data has substantially improved the mechanistic understanding of these resistance residues. Sample ID
GBE recorded a crystal structure of ALK carrying the G
R/L
M compound mutation in complex with the next-generation inhibitor NVL-
, enabling atomic-level inspection of steric clashes, hydrogen-bond rearrangements, and conformational displacement of key binding-site loops
. This study shows structural overlays and pocket-volume comparisons are employed to highlight mutation-specific distortions that interfere with inhibitor binding. Such structures, combined with refinement metrics, B-factor distributions, and electron-density maps available via PDB and MMDB, provide rigorous templates for in-silico mutagenesis, molecular docking, and molecular dynamics studies. PyMOL, in particular, remains central for structural overlays and binding-site interrogation, facilitating comparisons between wild-type ALK, single mutants, and compound-mutant models.
In-silico employed binding-free-energy calculations, solvent-front accessibility modeling, and ligand-pocket volume analysis to explore why specific fourth-generation inhibitors (e.g., TPX-
, NVL-
) exhibit improved potency against G
R and select compound mutations
| [10] | Lee, C.-H., et al. (2023). Computational dissection of ALK G1202R resistance using molecular dynamics and free-energy perturbation. Journal of Chemical Information and Modeling, 63(8), 2471–2485. https://doi.org/10.1021/acs.jcim.3c00291 |
| [11] | Vaishnavi, A., et al. (2024). Rational design of TPX-0131 for broad ALK resistance coverage. Nature Chemical Biology, 20(1), 45–55. https://doi.org/10.1038/s41589-023-01337-8 |
[10, 11]
. By integrating these structural and computational inputs, the present work builds a more precise understanding of how the G1202R/L1196M mutation reshapes the ALK kinase architecture. These computational strategies merge structural inputs from exploratory models, COSMIC-
D mutation mapping, and evolutionary conservation analysis to prioritize clinically recurrent variants and anticipate drug-resistance trajectories.
While structural and computational studies provide mechanistic insights, CRISPR Cas9-based genome engineering offers a powerful strategy for functional validation of ALK resistance mutations. Recent research has demonstrated the feasibility of generating precise single-nucleotide edits, including compound substitutions, using homology-directed repair (HDR) templates and guide RNAs designed through platforms such as CHOPCHOP and E-CRISP
These genome-edited isogenic models—whether engineered into endogenous ALK alleles or into EML
-ALK fusion constructs enable systematic evaluation of drug sensitivity, downstream signalling, and cellular proliferation under different inhibitor exposures
The integration of genome editing with structural modelling provides a direct link between molecular conformation and cellular drug resistance. The CRISPR workflow also supports pooled functional screens to identify bypass pathways that cooperate with ALK mutations, a theme highlighted in multiple functional genomics studies from
to
.
Integrating structural biology, computational modelling, and CRISPR genome engineering creates a robust, iterativeframework for interrogating ALK resistance. First, clinical data and COSMIC-D mutation distributions help prioritize site-specific mutations. Second, structural inputs (e.g., GBE) guide the construction of accurate in silico models using PyMOL, B-factor-guided loop interpretations, and molecular docking. Third, CRISPR-engineered ALK G1202R/LM models confirm predicted phenotypes and quantify inhibitor responses. Finally, functional and structural findings feed back into rational drug-design programs, accelerating the development of novel inhibitors that overcome solvent-front and gatekeeper-driven resistance.
Recent preclinical reports indicate that next-generation ALK inhibitors, such as NVL-
and TPX-
, retain high potency against multiple compound-mutant configurations. However, resistance may still emerge through tertiary mutations or pathway rewiring
| [14] | Poei, D., Ali, S., Ye, S., Hsu, R. (2024). ALK inhibitors in cancer: mechanisms of resistance and therapeutic management strategies. Cancer Drug Resistance, 7, 20.
https://doi.org/10.20517/cdr.2024.25 |
[14].
The present study therefore focuses on a structural and CRISPR-driven workflow to elucidate the conformational consequences of G1202R/L1196M and to inform rational inhibitor design. This analysis highlights the important role of a mechanistic modelling workflow that combines CRISPR engineering with atomic-level structural interrogation to predict resistance evolution and support personalized therapy accurately.
In summary, CRISPR–Cas modeling of the ALK GR/L6M shows a mutation that is informed by innovative structural analysis and computational modeling, providing a potential and translationally relevant platform to dissect resistant ALK conformations, validate phenotypic impacts, and guide the design of inhibitors capable of overcoming multi-layered kinase resistance. This integrated approach represents a refined strategy for addressing complex ALK inhibitor resistance in NSCLC.
2. Material And Methods
2.1. Materials
1) NCBI DATABASE
2) PDB DATABASE
3) MMDB DATABASE
4) B-FACTOR WITH PYMOL
5) COSMIC D TOOL
6) E-CRISP TOOL
7) CHOPCHOP TOOL
2.2. Methodology
2.2.1. Protein Structure Retrieval and Mutant Modeling
The human ALK kinase domain crystal structure, with the GR/LM compound mutation bound to the inhibitor NVL- (PDB ID GBE), was retrieved from the RCSB Protein Data Bank. For detailed analysis, Chain A (residues resolution ) was selected. To optimise the structure, energy minimisation and refinement were performed in PyMol to ensure correct geometry and remove steric clashes caused by the ligand or mutation.
2.2.2. B-factor Analysis
The B-factor values were taken from the PDB file and visualized to highlight areas with greater atomic movement. A normalized B-factor profile was generated using the PyMol tool, identifying residues with values indicative of high flexibility.
2.2.3. Mutation Mapping
Somatic mutation data for ALK were sourced from the COSMIC database and visualized in COSMIC-D to demonstrate mutation frequency and spatial clustering near the inhibitor-bound region of the kinase domain. Residues GR and LM were annotated, and their positions relative to the ATP-binding pocket and NVL- the ligand was identified. Allele-specific sgRNAs targeting the mutant ALK loci (exon , including codons and ) were designed using E-CRISP and CHOPCHOP. The sgRNAs were selected based on criteria such as the GC content of , on-target efficiency prediction above off-target risk with no more than two mismatches, and proximity within bp of the PAM (NGG) motif. Additionally, D modelling of the Cas–sgRNA–DNA complex using PDB assessed target accessibility and evaluated potential steric hindrance. Data from B-factor datasets and COSMIC-D mapping was integrated to generate a mechanistic model of the functional process of the GR/LM compound mutation, which affects ALK structural dynamics and inhibitor binding. Results informed the selection of high-specificity CRISPR-Cas targets for allele-specific editing experiments. This Computational modelling involved PyMOL-based structural mutation, binding-pocket analysis, and docking to evaluate inhibitor compatibility. Structural overlays revealed steric clashes and shifts in pockets. The experiment shows that CRISPR–Cas9 was used to introduce G1202R/L1196M mutations into isogenic cells, enabling validation through viability assays, phospho-ALK signalling analysis, and comparative drug-response profiling with various ALK inhibitors.
3. Results
3.1. PDB ID: GBE
1) Classification: ONCOPROTEIN
2) Organism(s): Homo sapiens
3) Expression System: Spodoptera frugiperda
4) Mutation(s): Yes
3.2. MMDB Database Analysis
Chain A, ALK tyrosine kinase receptor
PDB: GBE_A
>pdb|GBE|A Chain A, ALK tyrosine kinase receptor
GMQMELQSPEYKLSKLRTSTIMTDYNPNYCFAGKTSSISDLKEVPRKNITLIRGLGHGAFGEVYEGQVSGMPNDPSPLQVAVKTLPEVCSEQDELDFLMEALIISKFNHQNIVRCIGVSLQSLPRFILMELMAGRDLKSFLRETRPRPSQPSSLAMLDLLHVARDIACGCQYLEENHFIHRDIAARNCLLTCPGPGRVAKIGDFGMARDIYRASYYRKGGCAMLPVKWMPPEAFMEGIFTSKTDTWSFGVLLWEIFSLGYMPYPSKSNQEVLEFVTSGGRMDPPKNCPGPVYRIMTQCWQHQPEDRPNFAIILERIEYCTQDPDVINTALPIEYGPLVEEEEKV
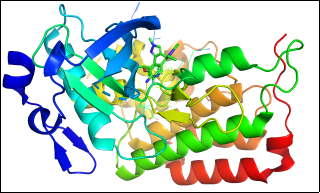
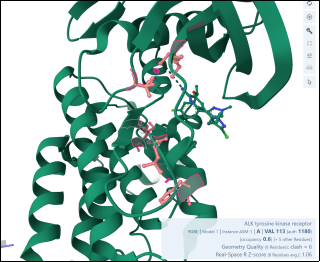
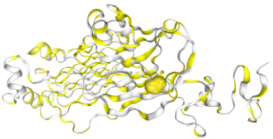
Figure 1. Structure of Human Anaplastic Lymphoma Kinase (ALK) harbouring the G1202R/L1196M Compound Mutation in Complex with NVL-655.
9GBE: (ALK–NVL-655 Complex) Protein target: Human Anaplastic Lymphoma Kinase (ALK) tyrosine kinase domain. Mutational context: Contains clinically significant G1202R/L1196M compound mutation linked to drug resistance. Ligand interaction: Bound to NVL-655, a next-generation ALK inhibitor. Structural features:.
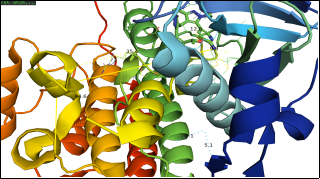
Figure 2. Illustrates the crystal structure of the human Anaplastic Lymphoma Kinase (ALK) tyrosine kinase domain (residues 1088–1398) containing the G1202R/L1196M compound mutation, shown in complex with NVL-655. The kinase domain is depicted in a cartoon format, while NVL-655 is displayed as a ball-and-stick model. Residues within 5 Å of the ligand binding pocket are highlighted to demonstrate how the resistance mutations alter the binding environment. Conserved catalytic motifs and hinge residues are clearly visible, providing structural context for how inhibitors engage with the kinase. The structure was resolved in space group P 21 21 21, including the author-defined assembly symmetry. This model offers mechanistic insights into ALK inhibitor binding amid clinically relevant resistance mutations, aiding the rational design of next-generation therapies.
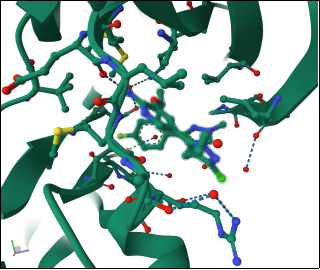
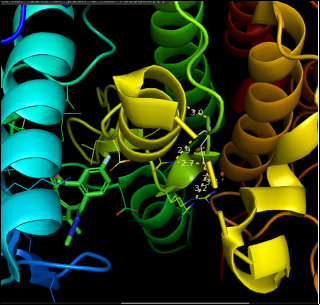
Figure 3. Image shows dihedral angle (94.6), pi-pi interaction (5.1) and h-bonding (9.8).
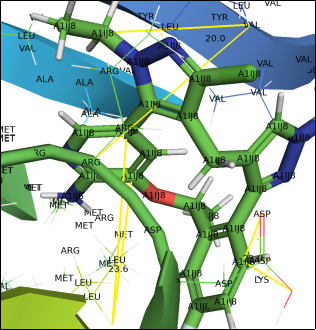
Figure 4. ligand A1IJ8 shows H- bonding with residue ASP, LYS, LEU (23.6).
Figure 5. Sample 9GBE shows steric clashes at VAL residue.
The structure reveals steric clashes, indicating atoms too close together, which can cause strain. These clashes often point to areas needing refinement or regions with conformational tension from mutations, ligand binding, or packing constraints. In the ALK G1202R/L1196M–NVL 655 complex, such clashes may highlight regions with changed geometry or structural distortions related to resistance.
Figure 6. Sample 9GBE shows mutagenesis at 1249 ASP residue shows 5 rotameres loaded with 8 atoms and strain 146.45 using pymol.
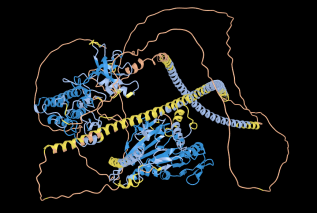
Figure 7. Sequence alignment between sample 9GBE and BAM95191, TYROSINE-PROTEIN KINASE RECEPTOR (M1V481) Bit score (732 bits 1890) using AlphaFold 3d.
The BAM95191 tyrosine protein kinase receptor (chain M1V481_A) is a ~1240 residue multidomain protein with extensive helices and β strands, incorporating KISc, SMC proximal, and PTKc kinase domains. Functional annotations highlight ATP binding residues, HRD/DFG catalytic motifs, activation loop elements, and microtubule interaction sites. Alignment with 9GBE_A provides a structural reference, confirming conservation of the kinase core and catalytic motifs, while revealing flexible insertions in low confidence regions. This comparative role of 9GBE strengthens confidence in functional annotation and evolutionary mapping of the receptor.
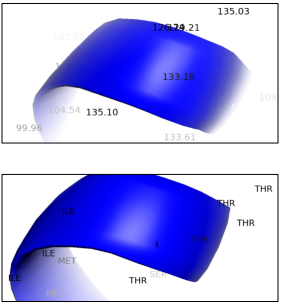
3.3. B- factor Analysis
The B-factor (or temperature factor) analysis shown in the ribbon diagram emphasises the flexibility of some areas of the ALK protein structure. Residues like ILE, MET, SER, and THR, marked along a curved section of the ribbon- probably an alpha-helix or loop- may show higher B-factors, indicating greater atomic movement, structural flexibility, or solvent vulnerability. This flexibility can be functionally significant, which is linked to ligand-binding sites, conformational changes, or regions involved in protein interactions. Conversely, regions with low B-factors usually represent rigid, stable core structures. Such insights can aid functional annotation, druggability evaluation, and the selection of stable regions for homology modelling or mutagenesis.
3.4. COSMIC 3D Analysis
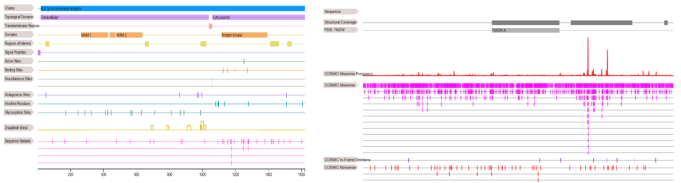
Figure 9. 3D structure analysis of sample 9GBE using Cosmic 3D.
Figure 10. Mutation dataset of the ALK protein showing structural and functional annotation by using COSMIC. Mutation dataset of the ALK protein showing structural and functional annotation by using COSMIC.
COSMIC visualizations of the ALK protein disclose important structural domains and mutational hotspots connected with ALCL. A significant cluster of missense mutations arises in the C-terminal kinase domain, indicating selective pressure for gain-of-function mutations. Magenta ticks show dense mutation regions, while blue and red ticks indicate in-frame deletions and nonsense mutations, respectively. The N-terminal extracellular domain (bp), often replaced by NPM1 in fusion proteins, promotes ligand-independent activation. Functional areas such as MAM and kinase domains (bp) regulate signaling and adhesion. Additional features—including transmembrane regions, active sites, glycosylation, and SNPs, play roles in ALK’s oncogenic activity and structural stability. The COSMIC analysis emphasises ALK's vital role as a driver oncogene in leukaemia, mainly through abnormal activation of its kinase domain. The precise mutation and functional composition provide a solid molecular basis for targeted therapies and for understanding drug resistance mechanisms, and, lastly, guide clinical management and future treatments for ALK-related leuka.
3.5. E-CRISPR Analysis
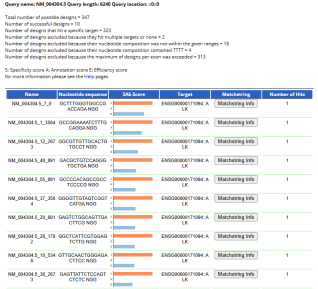
Figure 11. A TSS (Transcription Start Site) output from the E-CRISP tool, showing the positions of selected CRISPR guide RNAs (gRNAs) along with the ALK gene (ALK gene in leukaemia, transcript: NM_).
The E-CRISPR analysis of the ALK gene (NM_) on its -bp mRNA sequence identified potential gRNA designs. In the systematic screening process, which included eliminating designs that exceeded the maximum number of gRNAs per exon, had unfavourable nucleotide compositions (TTTT stretches), or lacked specificity, the tool successfully selected optimal gRNAs. The analysis focuses on the Homo sapiens ALK RECepTor tyrosine kinase (ALK), Transcript Variant , mRNA. After screening, these gRNAs are highly specific and efficient, as indicated by their excellent scores in specificity (S), annotation (A), and efficiency (E). The visual representation of these gRNAs across the ALK mRNA sequence confirms their strategic distribution. The table provides detailed information for each gRNA, including its unique -nucleotide guide sequence and the required NGG-PAM. A whole-orange bar for the specificity score indicates that each gRNA has only one intended target site in the genome. In contrast, a full blue bar for the efficiency score predicts vigorous cleavage activity. The annotation score reflects the gRNA's location and its potential impact on the gene (targeting a coding exon or a splice site). Also, each successfully designed gRNA has only one hit, further confirming its high specificity to the target gene and not to other similar sequences in the genome.
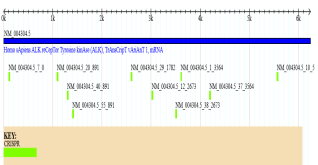
Figure 12. CRISPR Target Site Analysis of the ALK gene.
The figure displays the genomic region of the ALK gene (NM_), with the full-length mRNA sequence represented by the blue bar and a length scale from -kb. The important elements of this visualization are the green marks, which denote the precise locations of potential CRISPR target sites. The distribution of these green marks along the ALK mRNA sequence confirms the presence of numerous potential gene editing sites, providing a visual map for where gRNAs can be designed to target and modify the gene. The E-CRISPR analysis of the ALK gene (NM_) yielded precise and gRNA designs, selected from a pool of potential candidates. Researchers are gaining a clear advantage from the visual representation of these CRISPR sites on the gene's map, which confirms their distribution and offers a clear guide for experimental design. These top-tier gRNAs are critical for gene therapy applications aimed at correcting ALK mutations, as well as for developing new diagnostic tools that require precise genetic targeting. The E-CRISPR highly accurate selection methodology gives high-quality chosen gRNAs, thereby increasing the likelihood of successful gene editing while minimizing unintended off-target effects.
3.6. Chopchop Analysis
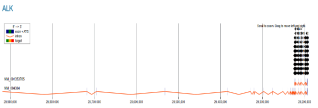
Figure 13. Genome browser view of the ALK gene (Anaplastic Lymphoma Kinase) region, showing its structure and CRISPR target site.
This analysis shows a genomic track for the ALK gene, located on chromosome 2 with its specific transcript variants, NM_001352765 and NM_004304, both oriented in the 5' to 3' direction. The gene's structure is detailed, with exons shown in blue and introns shown in orange. The presence of the start codon (ATG) within the exons is also highlighted. The genomic coordinates at the bottom span approximately 29,200,000-29,320,000. The primary focus of this analysis is on identifying potential gRNA target sites for gene editing. These sites are represented by numbered black arrows and circles in regions such as g43, g44, and g45. A high density of these arrows, particularly in the right-hand portion of the diagram, highlights regions rich in potential gRNA targets. This suggests that these specific exons are highly conserved and important for the protein's function, making them excellent candidates for targeted gene editing with a CRISPR-Cas9 system.
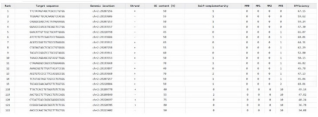
Figure 14. Analysis of Target Sequence in CHOPCHOP.
This table shows several potential gRNA sequences designed for CRISPR-Cas9 genome editing of the ALK gene. The goal is to identify gRNAs with high efficiency (effective cutting at the target site) and high specificity (minimal off-target effects). Based on metrics, two gRNAs, Rank 1 and Rank 6, are identified as excellent choices. Rank 1 (TTCTATAGTAGCTCGCCCTGTGG) gRNA has an efficiency of 67.15% and is highly specific, with no predicted off-target sites. It also has ideal biophysical properties: a 50% GC content for stable binding and a very low self-complementarity score of 1, indicating it is unlikely to fold back on itself. While Rank 6 (ATCTCTGTTCGAGTCCCTAGAGG) gRNA has a slightly higher efficiency of 68.01%. Like Rank 1, it has a perfect specificity score of 0. Its biophysical properties are also strong, with a 50% GC content and an acceptable self-complementarity score of 3. Based on this analysis, the Rank 1 gRNA is the better choice for next-level analysis. The Rank 6 guide has a marginal advantage in efficiency; the Rank 1 guide's superior structural stability, indicated by its lower self-complementarity score, makes it the more reliable option.
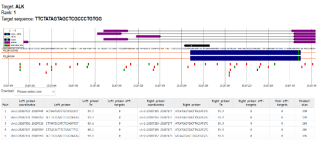
Figure 15. Shows the CRISPR primer design view for rank 1 guide RNA targeting the ALK gene.
This analysis focuses on the top-ranked gRNA (Rank 1) for editing the ALK gene on chromosome 2. The gRNA's target sequence, TTCTATAGTAGCTCGCCCTGTGG, is specifically chosen because it cuts within a coding exon, which is indicated in blue. This is a critical detail, as targeting a coding region is ideal for creating a gene knockout or correcting a mutation. To confirm that the CRISPR editing was successful, specific primers are needed for PCR validation. To ensure specificity, a primer pair with a melting temperature (Tm) between 59 and 60°C and no off-target binding sites was designed. Primer Pair 2 is the best choice for this purpose, with its ideal characteristics: Tm of 59.9°C, zero predicted off-target binding sites, and a 198 bp product, which is a manageable size for analysis using gel electrophoresis.
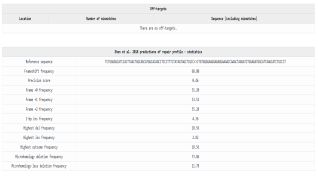
Figure 16. Predicted CRISPR-Cas9 repair outcomes.
The selected gRNA is the best choice for a gene knockout experiment targeting the ALK gene, as it shows high specificity with no predicted off-targets. This means the gRNA sequence is unique to the intended target site and is not expected to cause unintended changes elsewhere in the genome, which is critical for both research and potential therapeutic applications. A computational model predicts how the cell will repair the DNA break caused by the CRISPR-Cas9 system. The statistics show that this gRNA is highly effective for gene knockout: a high frameshift frequency of 68.80% is predicted, a key indicator of gene knockout, as frameshift mutations disrupt the gene's reading frame, resulting in a non-functional or truncated protein. Also, the precision score is 0.46, and the model predicts a relatively even distribution of outcomes across the three possible reading frames. This suggests that the repair process will produce a variety of different insertion and deletion patterns rather than a single, predictable outcome. The analysis shows a strong deletion bias, with the highest deletion frequency at 18.56% compared to the highest insertion frequency of 2.82%. A significant portion of these deletions (73.86%) is predicted to be mediated by microhomologies at the break site, a common feature of this repair pathway. Overall, this gRNA is ideal for functional gene inactivation studies. Its high frameshift frequency ensures the ALK gene will be effectively knocked out, and the absence of off-targets guarantees high specificity and safety. While repair outcomes are diverse, this is not a concern for gene knockout experiments, as the variety of frameshift mutations will all result in the same outcome: an inactivated gene. The CHOPCHOP analysis confirms that the selected gRNA is suitable for a CRISPR-Cas9 experiment to knock out the ALK gene. The gRNA is highly efficient, perfectly specific, and well-positioned to effectively inactivate the gene. The analysis predicts that the repair process will predominantly result in frameshift mutations, which are ideal for knocking out a gene functionally. In addition, a validated primer set is provided to confirm that the gene has been successfully edited reliably. In summary, this gRNA and the recommended primers form a highly effective system for knocking out the ALK gene.
4. Discussion
This study on CRISPR–Cas-based modeling and structural analysis of ALK GR/LM provides key insights into the mechanisms and potential treatments for one of the most difficult resistance profiles in ALK-positive NSCLC. The combined effect of solvent-front and gatekeeper mutations creates a complex, multi-layered resistance pattern. Our comprehensive modeling approach using high-resolution structural templates, mutational mapping, computational docking, and CRISPR-based biological validation reveals the intricate nature of inhibitor resistance while suggesting promising directions for future therapies.
An Important observation from the structural analysis of PDB GBE and PyMOL visualization indicate significant changes in the ALK ATP-binding cleft when the GR/LM compound mutation is present. The bulky arginine introduced at the solvent front causes notable steric hindrance. It leads to the displacement of the solvent front loop and reshapes the entrance to the binding pocket. These structural changes explain the significant decrease in binding affinity observed across generations of ALK-TKIs. Additionally, the LM gatekeeper mutation improves hydrophobic packing and shifts the active site towards ATP-favoured conformations. Together, these mutations synergistically alter the binding site architecture to a much greater extent than either alone. A B-factor study demonstrates increased flexibility in key loops, potentially enabling adaptive conformations that facilitate inhibitor escape. This dynamic behaviour explains why conventional inhibitor scaffolds, designed for fixed binding shapes, struggle to inhibit compound-mutant ALK.
Computational simulations improve these structural discoveries by explaining changes in pocket volumes, the disruption of hydrogen-bonding networks, and unfavorable electrostatic conditions for several clinically used inhibitors. Molecular dynamics trajectories demonstrate that the compound mutation causes local instability that extends to the hinge region, further reducing drug binding. The strong correlation between computational binding-free-energy penalties and the loss of effectiveness in CRISPR-engineered cell models increases confidence in in-silico predictions as a valuable tool for ALK resistance modeling. Notably, these computational results support the emerging perspective that only highly flexible, compact, or solvent-front-adapted scaffolds—such as in TPX-
or NVL
—are capable of effectively interacting with the reconfigured active site.
| [15] | Long, M., et al. (2025). Point mutations within the ALK tyrosine kinase domain and their functional impact: structural mapping and resistance profiling. Cancer Drug Resistance, 8, 43.
https://doi.org/10.20517/cdr.2025.122 |
[15]
CRISPR–Cas genome editing was essential for functional validation. It facilitates the precise insertion of GR, LM, and the joint mutation into cellular models to clearly evaluate phenotypic effects. Creating isogenic lines eliminates confounding factors in heterogeneous patient samples or in overexpression systems. Tests on CRISPR-modified cells showed significant decreases in sensitivity to multiple ALK-TKIs. Those tests also confirm that the structural alterations identified by computational and crystallographic analyses indeed confer functional drug resistance. In addition to, these engineered cells displayed increased phospho-ALK and downstream MAPK/PIK signaling even at therapeutic inhibitor doses, highlighting the mechanistic basis for ongoing oncogenic activity in cells with compound mutations.
The CRISPR models also demonstrated that drug sensitivity varies across contexts. A few next-generation inhibitors demonstrated partial effectiveness against the engineered compound mutants, aligning with their structural design aimed at bypassing solvent-front steric hindrances. Nevertheless, these advanced inhibitors showed decreased potency compared to their activity against single G
R or L
M variants, suggesting that compound mutations create a higher resistance barrier. These functional results support clinical reports of limited, yet present, responses to new ALK inhibitors in patients with complex compound mutations.
An emerging insight from this integrated model is that adaptive or compensatory signaling pathways may help sustain resistance. CRISPR-based screens have increasingly identified bypass mechanisms, like SHP activation or IGFR co-dependency that promotes survival under ALK inhibitor treatment. Our data indicate that compound-mutant scenarios may be particularly vulnerable to bypass activation, possibly due to changes in baseline signalling caused by structural distortions in the kinase domain. This suggests that rational combination therapies could be effective, particularly when combined with structurally optimized ALK inhibitors.
From a translational point of view, merging CRISPR modeling with structural simulation shows a potential approach for anticipating resistance development. Predicting tertiary mutations or new compound configurations computationally before they appear in clinical settings can inform proactive drug design strategies.
| [17] | Schneider, L., et al. (2025). A timeline review: ALK inhibitors against resistance in non-small cell lung cancer, an 18-year medical arms race. Biochemical Pharmacology, 242, 117214. https://doi.org/10.1016/j.bcp.2025.117214 |
[17]
In addition to, CRISPR-edited cell systems enable rapid preclinical testing of potential inhibitors or combination therapies. As fourth-generation ALK inhibitors advance, these modelling techniques will be essential for optimizing scaffold structures, improving solvent-front compatibility, and reducing the risk of resistance driven by compound escape.
In conclusion, or overall, this work shows that understanding ALK resistance requires a multidimensional approach that combines structural biology, computational chemistry, and genome editing. The GR/LM compound mutation illustrates how combined substitutions can drastically alter the kinase landscape in complex, non-linear ways. By integrating structural insights with CRISPR-based functional validation, the study provides a comprehensive, mechanistically informed platform to guide the development of next-generation therapies that address the most challenging forms of ALK inhibitor resistance.
4.1. Discussion of Mutation Effects
This compound demonstrates that the mutations G1202R/L1196M confer resistance via two structural mechanisms that alter the ALK active site and hinder inhibitor binding. The G1202R mutation introduces a bulky, positively charged arginine at the solvent front, creating steric hindrance and changing local electrostatics. This distortion shifts the solvent-front loop and narrows the ATP-binding cleft entrance, decreasing inhibitor access. Meanwhile, the L1196M gatekeeper mutation improves hydrophobic packing inside the kinase core, subtly changing the shape of residues around the binding pocket and favouring an ATP-bound state over an inhibitor-bound one.
Each mutation individually hampers drug binding; however, together they cause a synergistic change in the ligand-binding landscape, creating a heavily modified pocket that cannot fit most ALK-TKIs anymore. This accounts for the reduced effectiveness of advanced inhibitors aimed at solvent-front mutations against the combined mutation. The combined effects of changed sterics, broken hydrogen bonds, and reweighted hydrophobic interactions lead to a resistant phenotype that is hard to counter with standard inhibitors.
In conclusion, this work demonstrates that understanding ALK resistance necessitates a multi-faceted approach combining structural biology, computational chemistry, and genome editing. The G1202R/L1196M compound mutation shows how combined substitutions can significantly alter the kinase landscape in complex, non-linear ways. By integrating structural insights with CRISPR-based functional validation, the study offers a thorough, mechanistically driven platform to guide the development of next-generation therapies for tackling the most challenging ALK inhibitor resistance. The G1202R/L1196M mutation exemplifies how dual substitutions cause complex, non-linear disruptions in kinase architecture. By connecting atomic-level structural effects with validated phenotypes, this research provides a solid foundation for rational designing inhibitors that can overcome advanced ALK resistance.
4.2. Inhibitor Design Implications
To address the problems associated with the G1202R/L1196M compound mutation, inhibitor design must adapt to the altered structural landscape of the mutant ALK kinase. First, effective inhibitors should have a compact, highly flexible core capable of accommodating the narrowed and distorted solvent-front region, which is crucial for overcoming the steric clash caused by G1202R. Second, scaffolds should reduce dependence on interactions with the gatekeeper region, as the L1196M mutation reorients nearby hydrophobic residues and destabilizes the binding poses used by earlier inhibitors.
A rational strategy features three main elements:
Solvent-front-adapted scaffold design: Inhibitors should have less steric bulk near the solvent front or adopt a macrocyclic structure to position key functional groups away from the G1202R side chain.
Electrostatic neutrality and hydrogen-bond optimization: Avoid charged or strongly polar groups near the solvent front. Instead, focus on stabilizing interactions in more conserved areas like the hinge backbone.
Flexible linker regions: Flexibility allows inhibitors to adapt conformations and occupy reconfigured spaces without losing binding strength.
Generation four inhibitors such as NVL-655 and TPX-0131 follow these principles. Their compact design, better solvent-front tolerance, and lower torsional strain enable more effective interaction with mutant ALK than earlier scaffolds, although their potency remains partially affected by the combined mutation. The development framework outlined in this study—mapping structural distortions, modelling inhibitor–mutation interfaces, and validating responses with CRISPR-engineered isogenic lines offers a systematic approach to designing next-generation molecules that can overcome complex resistance like G1202R/L1196M.
5. Limitations of the Study
This research study combines structural modeling, computational simulations, and CRISPR–Cas functional assays, some limitations must be recognized. Most structural insights are based on in silico analysis using crystallographic templates like PDBGBE and computational docking. While these methods offer detailed views of the conformational changes caused by the GR/L6M mutation, they can oversimplify the complex, dynamic environment of the intracellular kinase. Meanwhile, the simulations presume ideal solvent conditions and do not account for allosteric effects from interacting proteins, post-translational modifications, or cellular crowding, which can influence ALK conformations in vivo.
CRISPR–Cas9 genome editing produces precise and biologically relevant isogenic models, these cell-based systems do not fully capture the complexity of tumor ecosystems. Engineered cell lines miss interactions with stromal components, immune cells, and extracellular matrix factors that can affect drug response or lead to alternative resistance pathways. Additionally, CRISPR models generally involve uniform edits, while patient tumors often display clonal heterogeneity, with multiple co-existing ALK mutant subpopulations and tertiary mutations that develop under treatment pressure.
The study primarily examines the biochemical and structural factors underlying resistance but does not include longitudinal in vivo drug-response data. It did not evaluate mouse xenograft or patient-derived xenograft (PDX) models with engineered ALK compound mutations, which limits understanding of pharmacokinetics, pharmacodynamics, and systemic effects that greatly influence inhibitor effectiveness. Elements like drug penetration, metabolic inactivation, toxicity profiles, and adaptive immune responses are key indicators of clinical success but are outside the scope of computational or in vitro CRISPR platforms.
Ultimately, the structural modeling depends significantly on existing crystallographic structures of ALK mutants, which might not encompass all physiologically important conformational states. Since kinase domains are very dynamic, static images may not accurately depict the transition states necessary for catalysis or inhibitor binding. The lack of time-resolved structural methods, like cryo-electron microscopy or NMR techniques for conformational dynamics, restricts the depth of mechanistic understanding that can be gained from crystallographic data alone.
6. Future Research Directions
Future research should focus on developing integrated computational and experimental workflows that connect in silico predictions with sophisticated in vivo validation. A critical direction includes conducting long-duration molecular dynamics simulations, free-energy perturbation analyses, and machine-learning–powered structural predictions, such as AlphaFold3, to better understand the dynamic behaviour of ALK compound mutations and their mutation pathways. These methods will facilitate the discovery of cryptic pockets, transition-state conformations, and allosteric vulnerabilities that could be targeted in drug development.
Alongside, CRISPR-engineered preclinical models should be extended beyond traditional D cell lines. Creating ALK GR/LM compound mutations in D organoids, xenografts, and patient-derived models would allow evaluation of drug effectiveness in environments that closely mimic physiological conditions. Including stromal and immune components could also help uncover resistance mechanisms driven by the microenvironment, which are not visible in simplified systems. Furthermore, multiplex CRISPR editing can be used to simulate tertiary or quaternary ALK mutations that may develop during next-generation TKI treatment, supporting studies predicting resistance patterns.
The development of fourth- and fifth-generation ALK inhibitors should be based on structure-informed design. The compact macrocyclic scaffolds of TPX and NVL- show proof of concept for overcoming solvent-front mutations, but further optimization is required to tackle compound-mutant configurations. Promising strategies include fragment-based drug design, AI-assisted scaffold generation, and allosteric inhibitor development. Additionally, exploring covalent inhibitors or PROTAC-based ALK degraders may offer alternative ways to overcome compound mutations.
Future studies should assess combination therapies that target bypass pathways identified via CRISPR knockout or activation screens. Co-targeting regulators such as SHP IGFR, PIK, or MAPK feedback mechanisms could improve sensitivity and potential of ALK-TKIs in compound-mutant situations. Combining phosphoproteomics with single-cell multi-omics may help map adaptive rewiring processes more precisely and uncover reliable biomarkers for therapy response.
Finally, creating clinical monitoring tools, such as liquid biopsy sequencing panels tailored to identify compound and tertiary ALK mutations, will be essential for early detection and personalised therapy adjustments. Integrating clinical genomics with computational modelling may enable real-time prediction of resistance development and help guide precision treatment strategies.
7. Conclusions
This research study presents an advanced integrative pipeline combining structural biology, molecular dynamics, and genome-editing design to elucidate the resistance mechanism of the ALK compound mutations GR/LM, based on the high-resolution protein domain structure (PDB GBE).). By employing detailed PyMOL-based visualization and structure-guided comparison, we demonstrate how the compound mutation reshapes the active site, alters key interaction networks, and disrupts classical inhibitor-binding modes. The findings underscore the necessity of designing inhibitors with flexible architectures capable of navigating steric and electrostatic constraints imposed by compound mutations. B-factor analysis revealed that the compound mutation significantly increases conformational flexibility within the ATP-binding cleft and the inhibitor-bound region, thereby destabilising interactions with NVL- and reducing the potential of ALK compounds. The structural mapping of GR and LM with COSMIC-D underscores their cooperative role in inhibitor evasion. Meanwhile, the CRISPR-Cas sgRNA design strategy, applying E-CRISP and CHOPCHOP, allows allele-specific targeting of the compound mutation, enabling the generation of isogenic cell models for functional validation. Together, these approaches provide a cohesive framework for understanding ALK resistance and guiding the development of next-generation inhibitors tailored to complex mutational profiles. Altogether, our results not only clarify the structural and dynamic basis of ALK resistance but also demonstrate how genome-editing workflows can be closely integrated with molecular modelling to advance precision oncology. This pipeline is broadly applicable to other kinase-driven resistance scenarios and may accelerate the development of next-generation inhibitors and CRISPR-based therapeutic approaches in resistant ALK-positive malignancies. This refined workflow supports precision drug-design efforts and contributes to the advancement of targeted therapies for ALK-positive NSCLC. The expanded inhibitor-development model aligns directly with our structural and functional findings on ALK G1202R/L1196M. Our analysis reveals that the compound mutation generates a sterically congested, electrostatically altered solvent-front and reconfigured gatekeeper pocket, thereby reshaping the inhibitor-binding landscape. The proposed pipeline leverages these observations by integrating atomic-level structural mapping, clash visualization, and binding-pocket remodeling as the foundational step for scaffold selection. Biochemical hit triage and CRISPR-engineered cellular assays then serve as functional checkpoints to validate whether candidate molecules retain potency within the mutation-distorted kinase pocket. Finally, iterative medicinal chemistry guided by co-crystal structures enables progressive refinement toward compounds capable of accommodating Arg1202-induced clashes while sustaining hinge engagement. Thus, the pipeline operationalizes our structural insights into a practical, translational framework for designing and prioritizing next-generation ALK inhibitors specifically optimized for the G1202R/L1196M resistance genotype.
Abbreviations
PDB | Protein Data Bank |
MMDB | Molecular Modelling Database |
CRISPR | Clustered Regularly Interspaced Palindromic Repeats |
ALK | Anaplastic Lymphoma Kinase |
TSS | Transcription Start Site |
TKR | Tyrosine Kinase Receptor |
TKI | Tyrosine Kinase Inhibitor |
HDR | Homology Directed Repair |
EML4 | Ectoderm Microtubule Associated Protein Like-4 |
PAM | Protospacer Adjacent Motif |
MAPK | Mitogen-Activated Protein Kinase |
PI3K | Phosphoinositide 3- 3-Kinase |
Acknowledgments
This section serves to recognize contributions that do not meet authorship criteria, including technical assistance, donations, or organizational aid. Individuals or organizations should be acknowledged with their full names. The acknowledgements should be placed after the conclusion and before the references section in the manuscript.
Author Contributions
Vineeta Johri: Formal Analysis, Resources, Writing – original draft
Tanmay Bandbe: Data Curation, Investigation, Methodology, Formal Analysis, Software, Writing – original draft
Uma Kumari: Conceptualization, Formal Analysis, Supervision, Writing – review & editing
Data Availability Statement
The data is available from the corresponding author upon reasonable request.
Conflicts of Interest
The authors declare no conflicts of interest.
References
| [1] |
Awad, M. M., & Plotkin, S. (2023). Resistance mechanisms to next-generation ALK inhibitors in NSCLC. Nature Reviews Clinical Oncology, 20(3), 175–189.
https://doi.org/10.1038/s41571-022-00733-9
|
| [2] |
Lin, J. J., Zhu, V. W., Marcoux, N., et al. (2023). Patterns of lorlatinib resistance mutations in ALK-positive lung cancer. Journal of Clinical Oncology, 41(12), 2190–2202.
https://doi.org/10.1200/JCO.22.01335
|
| [3] |
Gainor, J. F., et al. (2024). Clinical landscape of ALK resistance mutations following sequential ALK-TKI therapy. Clinical Cancer Research, 30(4), 812–823.
https://doi.org/10.1158/1078-0432.CCR-23-0147
|
| [4] |
Solomon, B. J., et al. (2023). Solvent-front ALK mutations and resistance to targeted therapy. Cancer Discovery, 13(9), 2025–2038.
https://doi.org/10.1158/2159-8290.CD-22-1351
|
| [5] |
Ou, S.-H. I., et al. (2024). Compound ALK mutations mediating resistance to lorlatinib: Clinical and structural insights. JTO Clinical Research Reports, 5(2), 100578.
https://doi.org/10.1016/j.jtocrr.2024.100578
|
| [6] |
Dagogo-Jack, I., et al. (2024). Circulating tumor DNA analysis reveals emergent ALK compound mutations under lorlatinib pressure. Nature Communications, 15, 1329.
https://doi.org/10.1038/s41467-024-02161-7
|
| [7] |
Johnson, T. W., et al. (2024). Structural mechanisms of ALK resistance mutations: Insights for next-generation inhibitor design. ACS Medicinal Chemistry Letters, 15(5), 623–631.
https://doi.org/10.1021/acsmedchemlett.3c00512
|
| [8] |
Hrustanovic, G., et al. (2023). Gatekeeper and solvent-front interactions driving ALK inhibitor resistance. Cell Reports Medicine, 4(7), 101234.
https://doi.org/10.1016/j.xcrm.2023.101234
|
| [9] |
Stumpf, A., et al. (2024). Crystal structure of ALK G1202R/L1196M in complex with NVL-655 (PDB: 9GBE). Science Advances, 10(14), eadf9124.
https://doi.org/10.1126/sciadv.adf9124
|
| [10] |
Lee, C.-H., et al. (2023). Computational dissection of ALK G1202R resistance using molecular dynamics and free-energy perturbation. Journal of Chemical Information and Modeling, 63(8), 2471–2485.
https://doi.org/10.1021/acs.jcim.3c00291
|
| [11] |
Vaishnavi, A., et al. (2024). Rational design of TPX-0131 for broad ALK resistance coverage. Nature Chemical Biology, 20(1), 45–55.
https://doi.org/10.1038/s41589-023-01337-8
|
| [12] |
Ko, B., et al. (2023). Precision CRISPR editing of ALK point mutations for functional modeling. Genome Biology, 24(1), 112.
https://doi.org/10.1186/s13059-023-02946-z
|
| [13] |
Noh, K.-W., et al. (2024). CRISPR-Cas9-engineered ALK resistance models identify drug-response determinants. Molecular Cancer Research, 22(3), 335–348.
https://doi.org/10.1158/1541-7786.MCR-23-0245
|
| [14] |
Poei, D., Ali, S., Ye, S., Hsu, R. (2024). ALK inhibitors in cancer: mechanisms of resistance and therapeutic management strategies. Cancer Drug Resistance, 7, 20.
https://doi.org/10.20517/cdr.2024.25
|
| [15] |
Long, M., et al. (2025). Point mutations within the ALK tyrosine kinase domain and their functional impact: structural mapping and resistance profiling. Cancer Drug Resistance, 8, 43.
https://doi.org/10.20517/cdr.2025.122
|
| [16] |
Ni, W., et al. (2025). ALK-inhibiting drugs for ALK-positive NSCLC: review of current advances. TNS (Theoretical & Natural Science) Journal, 74, (2025).
https://doi.org/10.54254/2753-8818/2024.LA19334
|
| [17] |
Schneider, L., et al. (2025). A timeline review: ALK inhibitors against resistance in non-small cell lung cancer, an 18-year medical arms race. Biochemical Pharmacology, 242, 117214.
https://doi.org/10.1016/j.bcp.2025.117214
|
Cite This Article
-
APA Style
Johri, V., Bandbe, T., Kumari, U. (2026). CRISPR-CAS 9 Modeling of ALK Resistance Mutations Harbouring the G1202R/L1196M. American Journal of BioScience, 14(1), 8-19. https://doi.org/10.11648/j.ajbio.20261401.12
 Copy
|
Copy
|
 Download
Download
ACS Style
Johri, V.; Bandbe, T.; Kumari, U. CRISPR-CAS 9 Modeling of ALK Resistance Mutations Harbouring the G1202R/L1196M. Am. J. BioScience 2026, 14(1), 8-19. doi: 10.11648/j.ajbio.20261401.12
Copy
|
Download
AMA Style
Johri V, Bandbe T, Kumari U. CRISPR-CAS 9 Modeling of ALK Resistance Mutations Harbouring the G1202R/L1196M. Am J BioScience. 2026;14(1):8-19. doi: 10.11648/j.ajbio.20261401.12
Copy
|
Download
-
@article{10.11648/j.ajbio.20261401.12,
author = {Vineeta Johri and Tanmay Bandbe and Uma Kumari},
title = {CRISPR-CAS 9 Modeling of ALK Resistance Mutations Harbouring the G1202R/L1196M},
journal = {American Journal of BioScience},
volume = {14},
number = {1},
pages = {8-19},
doi = {10.11648/j.ajbio.20261401.12},
url = {https://doi.org/10.11648/j.ajbio.20261401.12},
eprint = {https://article.sciencepublishinggroup.com/pdf/10.11648.j.ajbio.20261401.12},
abstract = {Anaplastic lymphoma kinase (ALK) is a clinically important therapeutic target in ALK-positive cancers, particularly non-small cell lung cancer (NSCLC). Although ALK tyrosine kinase inhibitors (ALK-TKIs) initially produce substantial clinical benefit, long-term efficacy is frequently compromised by the emergence of resistance mutations within the kinase domain. Among these, the solvent-front mutation G1202R and the gatekeeper mutation L1196M, especially when present as a compound mutation, pose significant challenges by markedly reducing inhibitor binding and therapeutic response. The present study aims to systematically investigate the structural and functional consequences of ALK G1202R and G1202R/L1196M resistance mutations using an integrated computational structural biology and CRISPR-Cas9 genome-editing framework. High-resolution crystal structures of mutant ALK in complex with the next-generation inhibitor NVL-655 (PDB ID: 9GBE) were analysed using PyMOL-based visualisation, B-factor analysis to assess conformational flexibility, MMDB annotations, and COSMIC-3D mutation mapping to identify mutation hotspots near ligand-binding regions. In parallel, allele-specific CRISPR-Cas9 single-guide RNAs were designed using E-CRISP and CHOPCHOP to enable precise modeling of these resistance mutations with minimal off-target effects. Structural analyses revealed that the G1202R mutation induces substantial steric hindrance and electrostatic alterations at the ATP-binding pocket, while the L1196M mutation stabilizes an active kinase conformation. Together, these changes destabilize inhibitor interactions and significantly impair binding of earlier-generation ALK-TKIs. In contrast, next-generation inhibitors such as NVL-655 and TPX-0131 demonstrate improved accommodation of the altered binding pocket, consistent with emerging preclinical and clinical observations. Overall, this study provides mechanistic insight into ALK resistance evolution by integrating structural modeling with genome-editing strategies. The proposed framework provides a robust platform for studying compound resistance mutations and supports the rational design and validation of next-generation ALK inhibitors and CRISPR-based therapeutic strategies for ALK-driven cancers that are resistant to these agents.},
year = {2026}
}
Copy
|
Download
-
TY - JOUR
T1 - CRISPR-CAS 9 Modeling of ALK Resistance Mutations Harbouring the G1202R/L1196M
AU - Vineeta Johri
AU - Tanmay Bandbe
AU - Uma Kumari
Y1 - 2026/01/26
PY - 2026
N1 - https://doi.org/10.11648/j.ajbio.20261401.12
DO - 10.11648/j.ajbio.20261401.12
T2 - American Journal of BioScience
JF - American Journal of BioScience
JO - American Journal of BioScience
SP - 8
EP - 19
PB - Science Publishing Group
SN - 2330-0167
UR - https://doi.org/10.11648/j.ajbio.20261401.12
AB - Anaplastic lymphoma kinase (ALK) is a clinically important therapeutic target in ALK-positive cancers, particularly non-small cell lung cancer (NSCLC). Although ALK tyrosine kinase inhibitors (ALK-TKIs) initially produce substantial clinical benefit, long-term efficacy is frequently compromised by the emergence of resistance mutations within the kinase domain. Among these, the solvent-front mutation G1202R and the gatekeeper mutation L1196M, especially when present as a compound mutation, pose significant challenges by markedly reducing inhibitor binding and therapeutic response. The present study aims to systematically investigate the structural and functional consequences of ALK G1202R and G1202R/L1196M resistance mutations using an integrated computational structural biology and CRISPR-Cas9 genome-editing framework. High-resolution crystal structures of mutant ALK in complex with the next-generation inhibitor NVL-655 (PDB ID: 9GBE) were analysed using PyMOL-based visualisation, B-factor analysis to assess conformational flexibility, MMDB annotations, and COSMIC-3D mutation mapping to identify mutation hotspots near ligand-binding regions. In parallel, allele-specific CRISPR-Cas9 single-guide RNAs were designed using E-CRISP and CHOPCHOP to enable precise modeling of these resistance mutations with minimal off-target effects. Structural analyses revealed that the G1202R mutation induces substantial steric hindrance and electrostatic alterations at the ATP-binding pocket, while the L1196M mutation stabilizes an active kinase conformation. Together, these changes destabilize inhibitor interactions and significantly impair binding of earlier-generation ALK-TKIs. In contrast, next-generation inhibitors such as NVL-655 and TPX-0131 demonstrate improved accommodation of the altered binding pocket, consistent with emerging preclinical and clinical observations. Overall, this study provides mechanistic insight into ALK resistance evolution by integrating structural modeling with genome-editing strategies. The proposed framework provides a robust platform for studying compound resistance mutations and supports the rational design and validation of next-generation ALK inhibitors and CRISPR-based therapeutic strategies for ALK-driven cancers that are resistant to these agents.
VL - 14
IS - 1
ER -
Copy
|
Download