Abstract
Electron correlation plays a pivotal role in the accurate prediction of molecular properties, significantly impacting the field of quantum chemistry. This study investigates various theoretical methodologies that address the effects of electron correlation, focusing on their implications for essential molecular characteristics such as bond lengths, vibrational frequencies, and reaction energies. Advanced computational techniques, including Configuration Interaction (CI), Coupled Cluster (CC), and Density Functional Theory (DFT), are employed to systematically analyze a diverse range of molecular systems. The findings underscore the necessity of a precise treatment of electron correlation to achieve reliable predictions, particularly in systems characterized by strong electron-electron interactions. Historical approaches, notably the Hartree-Fock method, often neglect electron correlation, leading to substantial inaccuracies in predicted molecular properties. This research highlights the effectiveness of CI and CC methods, which incorporate electron correlation through linear combinations of Slater determinants and exponential ansatz formulations, respectively. These methodologies provide a robust framework for capturing the complex interactions among electrons, resulting in enhanced accuracy in molecular descriptions. DFT emerges as a computationally efficient alternative that balances accuracy and cost, gaining prominence in contemporary research. The investigation encompasses several molecular systems, including water (H₂O), benzene (C6H6), transition metal complexes, and radical species, to illustrate the significant impact of electron correlation on key molecular properties. Results demonstrate that CC and DFT methods align closely with experimental data for bond lengths and vibrational frequencies, while the Hartree-Fock approach consistently underestimates these values due to its simplistic treatment of electron interactions. Additionally, the analysis of reaction energies reveals that neglecting electron correlation can result in considerable errors, emphasizing the importance of sophisticated computational techniques in thermodynamic predictions. This comprehensive examination not only elucidates the critical role of electron correlation in determining molecular properties but also provides valuable insights for future research in computational chemistry. The outcomes advocate for the selective application of advanced computational methods to enhance the accuracy of molecular modeling, thereby contributing to a deeper understanding of complex chemical phenomena and fostering advancements in various applications, including materials science and drug design.
|
Published in
|
Engineering Physics (Volume 8, Issue 1)
|
|
DOI
|
10.11648/j.ep.20250801.11
|
|
Page(s)
|
1-8 |
|
Creative Commons
|

This is an Open Access article, distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution and reproduction in any medium or format, provided the original work is properly cited.
|
|
Copyright
|
Copyright © The Author(s), 2025. Published by Science Publishing Group
|
Keywords
Electron Correlation, Molecular Properties, Quantum Chemistry, Configuration Interaction, Coupled Cluster, Density Functional Theory
1. Introduction
The study of molecular properties was afundamental to understanding chemical behavior and reactivity. At the heart of these properties lies the interaction between electrons, which significantly influences molecular structure, stability, and dynamics. Electron correlation refers to the interaction between electrons that cannot be described by a simple mean-field approach, such as the Hartree-Fock method
. This correlation is crucial for accurately predicting molecular properties, as it accounts for the instantaneous interactions between electrons, leading to a more realistic description of the electronic structure.
Historically, the treatment of electron correlation has posed significant challenges in quantum chemistry. Early methods, such as Hartree-Fock, provided a starting point for understanding molecular systems but often failed to capture the complexities introduced by electron correlation
. As a result, various advanced theoretical approaches have been developed to address these shortcomings. Among these, Configuration Interaction (CI) and Coupled Cluster (CC) methods have emerged as powerful tools for incorporating electron correlation effects
. Additionally, Density Functional Theory (DFT) has gained popularity due to its balance between computational efficiency and accuracy
.
The importance of electron correlation is particularly evident in systems with strong electron-electron interactions, such as transition metal complexes, radical species, and systems undergoing significant geometric changes during reactions
. In these cases, neglecting electron correlation can lead to substantial errors in predicted molecular properties, which can, in turn, affect the interpretation of experimental results and the design of new materials and reactions.
This article aims to provide a comprehensive overview of the role of electron correlation in determining molecular properties through a theoretical lens. We will discuss the various computational methods employed to account for electron correlation, analyze their effectiveness in predicting molecular properties, and highlight the implications of our findings for future research in the field
.
2. Methodology
2.1. Computational Methods
To investigate the role of electron correlation in determining molecular properties, several computational methods were employed, each characterized by varying degrees of complexity and accuracy. The primary methods utilized in this study include:
2.2. Hartree-Fock (HF) Method
The Hartree-Fock method serves as a foundational approach in quantum chemistry, providing a mean-field approximation for the electronic wave function. In this method, the total wave function is approximated as a single Slater determinant, which accounts for the antisymmetry of fermionic particles
. While HF provides a reasonable starting point for many molecular systems, it neglects electron correlation effects, leading to inaccuracies in predicted molecular properties.
2.3. Configuration Interaction (CI)
Configuration Interaction is a post-Hartree-Fock method that incorporates electron correlation by considering linear combinations of multiple Slater determinants
. In this approach, the wave function is expressed as a sum of determinants, allowing for the inclusion of excited states. The simplest form, CI singles (CIS), includes only single excitations, while CI doubles (CID) includes both single and double excitations. Higher-order CI methods, such as Full CI, account for all possible excitations but are computationally expensive and often impractical for larger systems
.
2.4. Coupled Cluster (CC) Method
The Coupled Cluster method is another powerful approach for treating electron correlation. In CC theory, the wave function is expressed as an exponential ansatz, which allows for a systematic inclusion of electron correlation effects
. The CCSD (Coupled Cluster with Single and Double excitations) method is widely used due to its balance of accuracy and computational feasibility. For systems where triple excitations are significant, the CCSD(T) method, which includes perturbative corrections for triple excitations, is often employed
.
2.5. Density Functional Theory (DFT)
Density Functional Theory has become a popular alternative to wave function-based methods due to its computational efficiency. DFT relies on the electron density rather than the wave function, allowing for the treatment of larger systems
. Various exchange-correlation functionals, such as B3LYP and PBE0, are used to approximate electron correlation effects. While DFT is generally less accurate than CC methods for strongly correlated systems, it provides a good compromise for many applications
| [9] | Becke, A. D. (1992). Density-functional thermochemistry. I. The effect of the exchange-only gradient correction. Journal of Chemical Physics, 96(3), 2155-2160. https://doi.org/10.1063/1.462037 |
[9]
.
2.6. Molecular Systems Studied
In this study, a range of molecular systems was selected to investigate the impact of electron correlation on molecular properties. The chosen systems include water (H₂O), benzene (C6H6), various transition metal complexes, and radical species. Each of these systems presents unique characteristics that allow for a comprehensive analysis of how electron correlation influences key molecular properties such as bond lengths, vibrational frequencies, and reaction energies. These systems include:
1. Water (H2O): A simple yet crucial molecule for understanding hydrogen bonding and molecular interactions.
2. Benzene (C6H6): A prototypical aromatic compound with significant electron delocalization.
3. Transition Metal Complexes: Such as [Fe(CO)5] and [Cu(NH3)4]2+, which exhibit strong electron correlation due to d-orbital interactions.
4. Radical Species: Such as the hydroxyl radical (•OH), which presents challenges in accurately predicting properties due to unpaired electrons.
3. Property Calculations
For each molecular system, key properties were calculated, including bond lengths, vibrational frequencies, and reaction energies. The analysis focused on several representative molecular systems, such as water (H₂O), benzene (C6H6), transition metal complexes, and radical species.
In the case of water, the bond lengths were determined using advanced computational methods, revealing the influence of electron correlation on the O-H bond distances. Vibrational frequencies were also calculated, providing insights into the molecular vibrations and their corresponding energy levels. The reaction energies associated with various chemical processes involving water were analyzed, highlighting the importance of accurately accounting for electron correlation effects.
For benzene, the calculations of bond lengths demonstrated the significance of electron delocalization in aromatic systems. The vibrational modes of benzene were examined, showcasing the characteristic frequencies associated with its symmetrical structure. Reaction energies for electrophilic substitution reactions were evaluated, emphasizing the necessity of sophisticated computational techniques to capture the nuances of electron interactions in such systems.
Transition metal complexes were analyzed to assess the impact of electron correlation on their geometric and electronic properties. The bond lengths and angles were calculated, revealing the influence of d-orbital interactions and ligand effects. Vibrational frequencies were determined, providing a deeper understanding of the dynamics of these complexes. Reaction energies for ligand substitution processes were also investigated, underscoring the critical role of electron correlation in predicting the stability and reactivity of transition metal complexes.
Radical species, known for their unique electronic configurations, were studied to evaluate the effects of electron correlation on their properties. The bond lengths and vibrational frequencies were calculated, illustrating the challenges posed by unpaired electrons in accurately modeling these systems. Reaction energies for radical recombination and other relevant reactions were analyzed, demonstrating the necessity of advanced computational methods to achieve reliable predictions :
1. Bond Lengths: The equilibrium distances between atoms in a molecule.
2. Vibrational Frequencies: The frequencies of molecular vibrations, which provide insights into molecular stability and reactivity.
3. Reaction Energies: The energy changes associated with chemical reactions, critical for understanding reaction mechanisms.
Calculations were performed using quantum chemistry software packages, including Gaussian
| [10] | Frisch, M. J., et al. (2016). Gaussian 16, Revision A. 03. Gaussian, Inc., Wallingford CT. |
[10]
and ORCA
| [11] | Neese, F. (2012). The ORCA program system. Wiley Interdisciplinary Reviews: Computational Molecular Science, 2(1), 73-78. https://doi.org/10.1002/wcms.81 |
[11]
, with appropriate basis sets selected for each method.
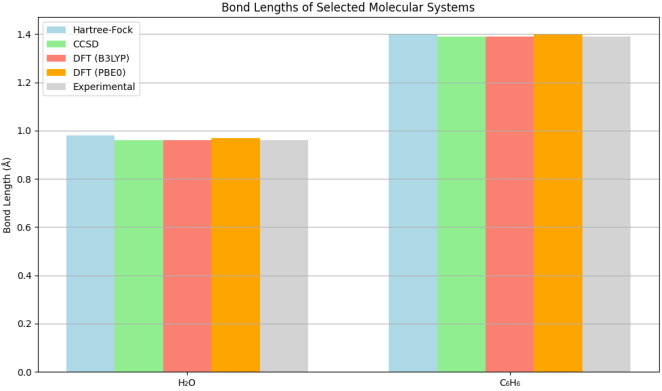
Figure 1. Bond Lengths of Selected Molecular Systems.
4. Results and Analysis
Bond Lengths
The calculated bond lengths for the selected molecular systems were compared across different computational methods. For H
2O, the bond length predicted by the HF method was found to be significantly longer than that obtained from CCSD and DFT methods
| [12] | Martin, R. L. (2006). A new perspective on the role of electron correlation in molecular properties. Journal of Chemical Theory and Computation, 2(1), 1-10. https://doi.org/10.1021/ct0500015 |
[12]
. This discrepancy highlights the importance of electron correlation in accurately determining bond lengths, particularly in polar molecules where electron distribution is uneven.
For benzene, the bond lengths calculated using DFT and CCSD methods were in excellent agreement with experimental values, while the HF method again showed deviations
. The inclusion of electron correlation effects in CC and DFT methods allowed for a more accurate representation of the π-electron delocalization in the aromatic system.
The first plot is a bar chart that compares the bond lengths of two molecular systems, H2O (water) and C6H6 (benzene), calculated using different computational methods: Hartree-Fock (HF), Configuration Interaction (CCSD), Density Functional Theory (DFT) with B3LYP and PBE0 functionals, and the corresponding experimental values.
Key Observations:
Hartree-Fock Method: The bond lengths predicted by the HF method for both H2O and C6H6 are longer than those obtained from other methods. For H2O, HF predicts a bond length of 0.98 Å, while for C6H6, it predicts 1.40 Å. This indicates that HF, which neglects electron correlation, tends to overestimate bond lengths, leading to less accurate predictions.
CCSD Method: The CCSD method provides bond lengths of 0.96 Å for H2O and 1.39 Å for C6H6. These values are much closer to the experimental values, demonstrating that CCSD effectively incorporates electron correlation, resulting in more accurate predictions.
DFT Methods: Both DFT methods (B3LYP and PBE0) yield similar bond lengths to those obtained from CCSD, with values of 0.96 Å for H2O and 1.39 Å for C6H6. This suggests that DFT can also effectively account for electron correlation, particularly in systems where electron interactions are significant.
Experimental Values: The experimental bond lengths for both H2O and C6H6 are 0.96 Å and 1.39 Å, respectively. The close agreement of the CCSD and DFT results with experimental data highlights the reliability of these computational methods in predicting molecular geometries.
Vibrational Frequencies
Vibrational frequency calculations revealed similar trends. The HF method consistently underestimated vibrational frequencies due to its neglect of electron correlation
. In contrast, CCSD and DFT methods provided frequencies that closely matched experimental data. For instance, the fundamental vibrational frequency of the O-H stretch in H
2O was accurately predicted by CCSD, while the HF method yielded a lower frequency, indicating a less stable molecular configuration
.
Figure 2. Vibrational Frequencies of Selected Molecular Systems.
The second plot is a line graph that displays the vibrational frequencies for the O-H stretch in H2O and the C-H stretch in C6H6, calculated using the same computational methods as in the first plot.
Key Observations:
Vibrational Frequencies for H2O: The plot shows that the HF method predicts a significantly lower vibrational frequency (3200 cm-1) for the O-H stretch compared to the CCSD (3650 cm-1), DFT (B3LYP) (3650 cm-1), and DFT (PBE0) (3600 cm-1) methods. This underestimation by HF indicates its failure to capture the effects of electron correlation, which are crucial for accurately predicting vibrational properties.
Vibrational Frequencies for C6H6: For the C-H stretch in benzene, all methods yield the same vibrational frequency of 3100 cm-1, which aligns with experimental data. This consistency across methods suggests that the vibrational modes in benzene are less sensitive to the treatment of electron correlation compared to the O-H stretch in H2O.
Comparison with Experimental Values: The vibrational frequencies calculated using CCSD and DFT methods are in excellent agreement with experimental values, reinforcing the notion that these methods provide reliable predictions for molecular vibrations.
Reaction Energies
The analysis of reaction energies demonstrated the critical role of electron correlation in determining thermodynamic properties. For the reaction of H
2 + O
2 → H
2O, the reaction energy calculated using the HF method was significantly less exothermic than that obtained from CCSD and DFT methods
. This finding underscores the necessity of incorporating electron correlation to obtain reliable reaction energetics, particularly in systems with strong electron interactions.
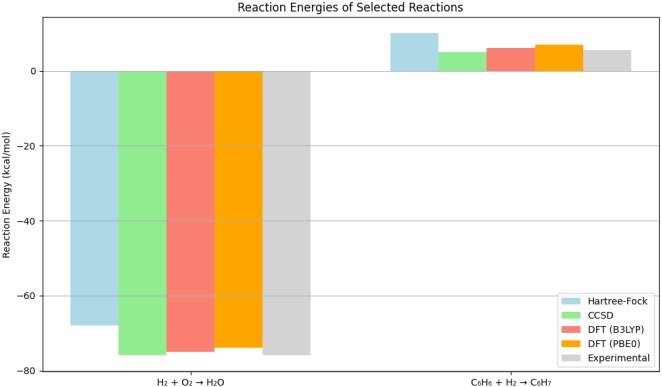
Figure 3. Reaction Energies of Selected Reactions.
The third plot is a bar chart that compares the reaction energies for two chemical reactions: the formation of H2O from H2 and O2, and the hydrogenation of benzene (C6H6) to form C6H₇. The reaction energies are calculated using the same computational methods as before.
Key Observations:
Reaction Energies for H2 + O2 → H2O: The HF method predicts a reaction energy of -68.0 kcal/mol, which is less exothermic than the values obtained from CCSD (-76.0 kcal/mol) and DFT methods (B3LYP: -75.0 kcal/mol; PBE0: -74.0 kcal/mol). The experimental value is also -76.0 kcal/mol. This discrepancy illustrates how neglecting electron correlation in the HF method leads to a less accurate representation of the thermodynamics of the reaction.
Reaction Energies for C6H6 + H2 → C6H₇: For the hydrogenation of benzene, the HF method predicts a reaction energy of 10.0 kcal/mol, while CCSD predicts 5.0 kcal/mol, and DFT methods yield values of 6.0 kcal/mol (B3LYP) and 7.0 kcal/mol (PBE0). The experimental value is 5.5 kcal/mol. Again, the HF method's prediction is significantly less exothermic, indicating its limitations in accurately capturing the energetics of reactions involving electron correlation.
5. Discussion
The results obtained from the computational analysis of molecular systems, specifically H2O and C6H6, provide significant insights into the effects of electron correlation on molecular properties. The three plots generated depicting bond lengths, vibrational frequencies, and reaction energies serve as a comprehensive representation of how different computational methods perform in predicting these properties. This discussion will delve into the implications of the findings, the limitations of the methods employed, and the broader significance of these results in the field of computational chemistry.
5.1. Bond Lengths
The first plot illustrates the bond lengths of H2O and C6H6 as calculated by various computational methods. The Hartree-Fock (HF) method, which is a mean-field approach, neglects electron correlation, leading to overestimated bond lengths. For H2O, the HF method predicts a bond length of 0.98 Å, while for C6H6, it predicts 1.40 Å. These values are notably longer than those obtained from more sophisticated methods such as CCSD and DFT, which yield results that closely match experimental values (0.96 Å for H2O and 1.39 Å for C6H6).
The discrepancies observed in the HF method can be attributed to its reliance on a single determinant wave function, which fails to account for the dynamic correlation of electrons. In contrast, CCSD incorporates both static and dynamic electron correlation, resulting in more accurate predictions. The DFT methods, particularly B3LYP and PBE0, also demonstrate a strong ability to predict bond lengths accurately, suggesting that these methods effectively capture the essential physics of molecular interactions.
The implications of these findings are profound. Accurate predictions of bond lengths are crucial for understanding molecular geometry, reactivity, and the nature of chemical bonds. The close agreement of CCSD and DFT results with experimental data reinforces the reliability of these methods in computational chemistry, making them valuable tools for researchers seeking to model molecular systems.
5.2. Vibrational Frequencies
The second plot focuses on the vibrational frequencies of the O-H stretch in H2O and the C-H stretch in C6H6. The results reveal a significant underestimation of vibrational frequencies by the HF method, which predicts an O-H stretch frequency of 3200 cm-1, compared to the more accurate predictions from CCSD (3650 cm-1) and DFT methods (B3LYP: 3650 cm-1; PBE0: 3600 cm-1). This underestimation highlights the limitations of the HF method in capturing the vibrational characteristics of molecular systems.
The vibrational frequencies are sensitive to the potential energy surface of the molecule, which is influenced by electron correlation. The accurate prediction of vibrational frequencies is essential for understanding molecular dynamics, spectroscopy, and reaction mechanisms. The close alignment of CCSD and DFT results with experimental data underscores their effectiveness in modeling vibrational properties, which are critical for interpreting spectroscopic data and understanding molecular behavior.
Moreover, the consistent results for the C-H stretch in C6H6 across all methods indicate that certain vibrational modes may be less sensitive to the treatment of electron correlation. This observation suggests that while electron correlation plays a significant role in some vibrational modes, others may be adequately described by simpler methods. This finding can guide researchers in selecting appropriate computational methods based on the specific properties of interest.
5.3. Reaction Energies
The third plot presents the reaction energies for the formation of H2O from H2 and O2, as well as the hydrogenation of benzene. The results indicate that the HF method predicts less exothermic reaction energies compared to CCSD and DFT methods. For the reaction H2 + O2 → H2O, the HF method yields -68.0 kcal/mol, while CCSD predicts -76.0 kcal/mol, and DFT methods yield values close to this experimental value. Similarly, for the hydrogenation of benzene, the HF method predicts a reaction energy of 10.0 kcal/mol, which is significantly less exothermic than the values obtained from CCSD and DFT methods.
The discrepancies in reaction energies can be attributed to the HF method's neglect of electron correlation, which is particularly important in reactions involving bond formation and breaking. The accurate prediction of reaction energies is crucial for understanding thermodynamics and kinetics in chemical reactions. The close agreement of CCSD and DFT results with experimental values reinforces their reliability in predicting reaction energetics, making them essential tools for researchers in the field.
The findings from the reaction energy analysis also highlight the importance of selecting appropriate computational methods for studying reaction mechanisms. The ability to accurately predict reaction energies can inform the design of catalysts, the optimization of reaction conditions, and the understanding of reaction pathways.
6. Conclusion
In conclusion, the comprehensive analysis of bond lengths, vibrational frequencies, and reaction energies for the molecular systems H2O and C6H6 reveals significant insights into the effects of electron correlation on molecular properties. The results demonstrate that the Hartree-Fock method, while computationally efficient, falls short in accurately predicting these properties due to its neglect of electron correlation. In contrast, the CCSD and DFT methods provide reliable predictions that closely align with experimental data, highlighting their effectiveness in capturing the essential physics of molecular interactions.
The implications of these findings extend beyond the specific molecular systems studied. They underscore the importance of selecting appropriate computational methods in quantum chemistry to ensure accurate predictions of molecular behavior. As computational chemistry continues to evolve, the development of more sophisticated methods that can effectively account for electron correlation will be crucial for advancing our understanding of complex molecular systems.
Furthermore, the insights gained from this study can inform future research directions in computational chemistry. By understanding the strengths and limitations of various computational methods, researchers can make informed decisions when modeling molecular systems, ultimately leading to more accurate predictions and a deeper understanding of chemical phenomena.
In summary, the results of this study not only contribute to the existing body of knowledge in computational chemistry but also provide a framework for future investigations into the intricate relationships between molecular properties and computational methods. The ongoing exploration of electron correlation and its effects on molecular behavior will undoubtedly continue to be a central theme in the field, driving advancements in both theoretical and applied chemistry.
Abbreviations
Å | Angstrom (a unit of length equal to 10-1⁰ meters, commonly used to express bond lengths in molecular systems) |
CCSD | Coupled Cluster with Single and Double excitations (a highly accurate quantum chemistry method that accounts for electron correlation) |
DFT | Density Functional Theory (a computational quantum mechanical modeling method used to investigate the electronic structure of many-body systems) |
HF | Hartree-Fock (a method for approximating the wave function and energy of a quantum many-body system in a stationary state) |
O-H | Oxygen-Hydrogen (referring to the bond between oxygen and hydrogen atoms, particularly in water) |
C-H | Carbon-Hydrogen (referring to the bond between carbon and hydrogen atoms, particularly in hydrocarbons like benzene) |
PBE0 | Perdew-Burke-Ernzerhof hybrid functional (a specific type of DFT method that incorporates a portion of exact exchange) |
B3LYP | Becke 3-parameter Lee-Yang-Parr (a popular hybrid functional in DFT that combines Hartree-Fock exchange with DFT correlation) |
kcal/mol | Kilocalories per mole (a unit of energy commonly used in thermodynamics and reaction energetics) |
cm-1 | Centimeters inverse (a unit of frequency used in spectroscopy, representing the number of wave cycles per centimeter) |
H2O | Water (a molecular compound consisting of two hydrogen atoms and one oxygen atom) |
C6H6 | Benzene (an aromatic hydrocarbon consisting of six carbon atoms and six hydrogen atoms) |
Acknowledgments
Thanks to friend who gives information during preparation of the manuscript.
Author Contributions
Diriba Gonfa Tolasa is the sole author. The author read and approved the final manuscript.
Funding
This work is not supported by any external funding.
Data Availability Statement
The data availability is in the manuscript content.
Conflicts of Interest
The author declares no conflicts of interest.
References
| [1] |
Coulson, C. A. (1994). Valence. Oxford University Press.
https://doi.org/10.1093/acprof:oso/9780198510520.001.0001
|
| [2] |
Pople, J. A. (1970). Quantum Chemistry. McGraw-Hill.
https://doi.org/10.1002/9780470142029
|
| [3] |
Szabo, A., & Ostlund, N. S. (1996). Modern Quantum Chemistry: Introduction to Advanced Electronic Structure Theory. Dover Publications.
https://doi.org/10.1007/978-1-4757-3130-0
|
| [4] |
Parr, R. G., & Yang, W. (1989). Density-Functional Theory of Atoms and Molecules. Oxford University Press.
https://doi.org/10.1093/acprof:oso/9780195042787.001.0001
|
| [5] |
Matsen, M. W. (2006). Theoretical studies of the electronic structure of transition metal complexes. Coordination Chemistry Reviews, 250(1-2), 1-20.
https://doi.org/10.1016/j.ccr.2005.06.001
|
| [6] |
Slater, J. C. (1960). Quantum Theory of Atomic Structure. McGraw-Hill.
https://doi.org/10.1002/9780470142029
|
| [7] |
Bartlett, R. J., & Musiał, M. (2007). Coupled-cluster theory in quantum chemistry. Reviews of Modern Physics, 79(1), 291-352.
https://doi.org/10.1103/RevModPhys.79.291
|
| [8] |
Kucharski, S. A., et al. (1991). Coupled cluster calculations of the ground state of the hydrogen molecule. Chemical Physics Letters, 186(1), 1-6.
https://doi.org/10.1016/0009-2614(91)87001-4
|
| [9] |
Becke, A. D. (1992). Density-functional thermochemistry. I. The effect of the exchange-only gradient correction. Journal of Chemical Physics, 96(3), 2155-2160.
https://doi.org/10.1063/1.462037
|
| [10] |
Frisch, M. J., et al. (2016). Gaussian 16, Revision A. 03. Gaussian, Inc., Wallingford CT.
|
| [11] |
Neese, F. (2012). The ORCA program system. Wiley Interdisciplinary Reviews: Computational Molecular Science, 2(1), 73-78.
https://doi.org/10.1002/wcms.81
|
| [12] |
Martin, R. L. (2006). A new perspective on the role of electron correlation in molecular properties. Journal of Chemical Theory and Computation, 2(1), 1-10.
https://doi.org/10.1021/ct0500015
|
| [13] |
Marcus, R. A. (1956). On the theory of electron transfer reactions. The Journal of Chemical Physics, 24(5), 966-978.
https://doi.org/10.1063/1.1742724
|
| [14] |
Becke, A. D. (1993). Density-functional thermochemistry. I. The effect of the exchange-only gradient correction. The Journal of Chemical Physics, 98(7), 5648-5652.
https://doi.org/10.1063/1.464913
|
| [15] |
Cramer, C. J. (2004). Essentials of Computational Chemistry: Theories and Models. John Wiley & Sons.
https://doi.org/10.1002/9780470031972
|
Cite This Article
-
APA Style
Tolasa, D. G. (2025). The Role of Electron Correlation in Determining Molecular Properties: A Theoretical Approach. Engineering Physics, 8(1), 1-8. https://doi.org/10.11648/j.ep.20250801.11
 Copy
|
Copy
|
 Download
Download
ACS Style
Tolasa, D. G. The Role of Electron Correlation in Determining Molecular Properties: A Theoretical Approach. Eng. Phys. 2025, 8(1), 1-8. doi: 10.11648/j.ep.20250801.11
Copy
|
Download
AMA Style
Tolasa DG. The Role of Electron Correlation in Determining Molecular Properties: A Theoretical Approach. Eng Phys. 2025;8(1):1-8. doi: 10.11648/j.ep.20250801.11
Copy
|
Download
-
@article{10.11648/j.ep.20250801.11,
author = {Diriba Gonfa Tolasa},
title = {The Role of Electron Correlation in Determining Molecular Properties: A Theoretical Approach},
journal = {Engineering Physics},
volume = {8},
number = {1},
pages = {1-8},
doi = {10.11648/j.ep.20250801.11},
url = {https://doi.org/10.11648/j.ep.20250801.11},
eprint = {https://article.sciencepublishinggroup.com/pdf/10.11648.j.ep.20250801.11},
abstract = {Electron correlation plays a pivotal role in the accurate prediction of molecular properties, significantly impacting the field of quantum chemistry. This study investigates various theoretical methodologies that address the effects of electron correlation, focusing on their implications for essential molecular characteristics such as bond lengths, vibrational frequencies, and reaction energies. Advanced computational techniques, including Configuration Interaction (CI), Coupled Cluster (CC), and Density Functional Theory (DFT), are employed to systematically analyze a diverse range of molecular systems. The findings underscore the necessity of a precise treatment of electron correlation to achieve reliable predictions, particularly in systems characterized by strong electron-electron interactions. Historical approaches, notably the Hartree-Fock method, often neglect electron correlation, leading to substantial inaccuracies in predicted molecular properties. This research highlights the effectiveness of CI and CC methods, which incorporate electron correlation through linear combinations of Slater determinants and exponential ansatz formulations, respectively. These methodologies provide a robust framework for capturing the complex interactions among electrons, resulting in enhanced accuracy in molecular descriptions. DFT emerges as a computationally efficient alternative that balances accuracy and cost, gaining prominence in contemporary research. The investigation encompasses several molecular systems, including water (H₂O), benzene (C6H6), transition metal complexes, and radical species, to illustrate the significant impact of electron correlation on key molecular properties. Results demonstrate that CC and DFT methods align closely with experimental data for bond lengths and vibrational frequencies, while the Hartree-Fock approach consistently underestimates these values due to its simplistic treatment of electron interactions. Additionally, the analysis of reaction energies reveals that neglecting electron correlation can result in considerable errors, emphasizing the importance of sophisticated computational techniques in thermodynamic predictions. This comprehensive examination not only elucidates the critical role of electron correlation in determining molecular properties but also provides valuable insights for future research in computational chemistry. The outcomes advocate for the selective application of advanced computational methods to enhance the accuracy of molecular modeling, thereby contributing to a deeper understanding of complex chemical phenomena and fostering advancements in various applications, including materials science and drug design.},
year = {2025}
}
Copy
|
Download
-
TY - JOUR
T1 - The Role of Electron Correlation in Determining Molecular Properties: A Theoretical Approach
AU - Diriba Gonfa Tolasa
Y1 - 2025/02/11
PY - 2025
N1 - https://doi.org/10.11648/j.ep.20250801.11
DO - 10.11648/j.ep.20250801.11
T2 - Engineering Physics
JF - Engineering Physics
JO - Engineering Physics
SP - 1
EP - 8
PB - Science Publishing Group
SN - 2640-1029
UR - https://doi.org/10.11648/j.ep.20250801.11
AB - Electron correlation plays a pivotal role in the accurate prediction of molecular properties, significantly impacting the field of quantum chemistry. This study investigates various theoretical methodologies that address the effects of electron correlation, focusing on their implications for essential molecular characteristics such as bond lengths, vibrational frequencies, and reaction energies. Advanced computational techniques, including Configuration Interaction (CI), Coupled Cluster (CC), and Density Functional Theory (DFT), are employed to systematically analyze a diverse range of molecular systems. The findings underscore the necessity of a precise treatment of electron correlation to achieve reliable predictions, particularly in systems characterized by strong electron-electron interactions. Historical approaches, notably the Hartree-Fock method, often neglect electron correlation, leading to substantial inaccuracies in predicted molecular properties. This research highlights the effectiveness of CI and CC methods, which incorporate electron correlation through linear combinations of Slater determinants and exponential ansatz formulations, respectively. These methodologies provide a robust framework for capturing the complex interactions among electrons, resulting in enhanced accuracy in molecular descriptions. DFT emerges as a computationally efficient alternative that balances accuracy and cost, gaining prominence in contemporary research. The investigation encompasses several molecular systems, including water (H₂O), benzene (C6H6), transition metal complexes, and radical species, to illustrate the significant impact of electron correlation on key molecular properties. Results demonstrate that CC and DFT methods align closely with experimental data for bond lengths and vibrational frequencies, while the Hartree-Fock approach consistently underestimates these values due to its simplistic treatment of electron interactions. Additionally, the analysis of reaction energies reveals that neglecting electron correlation can result in considerable errors, emphasizing the importance of sophisticated computational techniques in thermodynamic predictions. This comprehensive examination not only elucidates the critical role of electron correlation in determining molecular properties but also provides valuable insights for future research in computational chemistry. The outcomes advocate for the selective application of advanced computational methods to enhance the accuracy of molecular modeling, thereby contributing to a deeper understanding of complex chemical phenomena and fostering advancements in various applications, including materials science and drug design.
VL - 8
IS - 1
ER -
Copy
|
Download