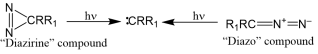
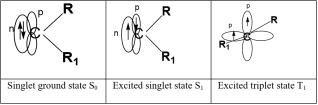
Carbenes are unstable biradical molecules with a longer lifetime in space than on Earth. This theoretical study focused on analyzing the spectroscopic properties of methylene and monohalogenated and dihalogenated derivatives in the ultraviolet-visible range. Structures of these compounds can exist in two electronic states depending on the orbitals containing the non-bonding electrons: singlet S0 and triplet T1. Three theoretical levels HF/6-311++G (d, p), MP2/6-311++G (d, p), and B3LYP/6-311++G (d, p) were used to perform this analysis. Absorption spectra were calculated for the optimized structures (S0 and T1) of methylene, three hydrohalogenocarbenes and two dihalogenocarbenes at each of the chosen theoretical levels. The calculations allowed us to study the behavior of these carbenes in ultraviolet-visible absorption. It was determined that, at maximum absorption, all of these carbenes in the singlet S0 state absorb in the visible range. However, in the T1 triplet state, they absorb in the ultraviolet range. Under maximum oscillation conditions, these carbenes absorb in the ultraviolet regardless of the electronic state of the structure. The calculated absorption wavelengths show that the transition from the ground state to their excited states of the studied carbenes is sometimes accompanied by a bathochromic effect and sometimes by a hypsochromic effect.
| Published in | American Journal of Quantum Chemistry and Molecular Spectroscopy (Volume 10, Issue 1) |
| DOI | 10.11648/j.ajqcms.20261001.14 |
| Page(s) | 35-41 |
| Creative Commons |
This is an Open Access article, distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution and reproduction in any medium or format, provided the original work is properly cited. |
| Copyright |
Copyright © The Author(s), 2026. Published by Science Publishing Group |
Carbene, Theory Level, Spectroscopy, Wavelength, Bathochrome, Hypsochrome
Methylene | Hydrohalogenocarbenes | Dihalogenocarbenes |
|---|---|---|
|
X = F or Cl or Br |
X1 = X2 = F or Cl |
Calculation |
|
|
|
| ||
|---|---|---|---|---|---|---|
method |
|
|
|
|
|
|
HF | 125 | 126 | 0.20 | 0.08 | -1 | 0.12 |
B3LYP | 125 | 129 | 0.20 | 0.08 | - 4 | 0.12 |
MP2 | 125 | 127 | 0.20 | 0.08 | -2 | 0.12 |
Calculation |
|
|
|
| ||
|---|---|---|---|---|---|---|
method |
|
|
|
|
| |
HF | 790 | 172 | 0.009 | 0.040 | 619 | -0.031 |
B3LYP | 742 | 173 | 0.009 | 0.037 | 569 | -0.028 |
MP2 | 749 | 172 | 0.009 | 0.039 | 577 | -0.030 |
Hydrohalo-genocarbenes |
|
|
|
| ||
|---|---|---|---|---|---|---|
|
|
|
|
|
| |
HF/6-311++G(d,p) | ||||||
CHF | 104 | 123 | 0.262 | 0.063 | 19 | 0.2 |
CHCl | 151 | 158 | 0.219 | 0.051 | 7 | 0.2 |
CHBr | 142 | 155 | 0.268 | 0.047 | 14 | 0.2 |
B3LYP /6-311++G(d,p) | ||||||
CHF | 104 | 124 | 0.256 | 0.062 | 20 | 0.2 |
CHCl | 152 | 157 | 0.208 | 0.051 | 6 | 0.2 |
CHBr | 141 | 180 | 0.307 | 0.362 | 39 | -0.1 |
MP2 /6-311++G(d,p) | ||||||
CHF | 104 | 124 | 0.259 | 0.061 | 20 | 0.2 |
CHCl | 149 | 157 | 0.229 | 0.052 | 8 | 0.2 |
CHBr | 142 | 155 | 0.232 | 0.069 | 14 | 0.2 |
Hydrohalo-genocarbenes |
|
|
|
| ||
|---|---|---|---|---|---|---|
|
|
|
|
|
| |
HF/6-311++G(d,p) | ||||||
CHF | 479 | 174 | 0.017 | 0.032 | -305 | -0.01 |
CHCl | 598 | 204 | 0.010 | 0.000 | -394 | 0.01 |
CHBr | 625 | 239 | 0.008 | 0.000 | -386 | 0.01 |
B3LYP /6-311++G(d,p) | ||||||
CHF | 470 | 172 | 0.016 | 0.030 | -298 | -0.01 |
CHCl | 580 | 202 | 0.010 | 0.000 | -377 | 0.01 |
CHBr | 601 | 236 | 0.008 | 0.000 | -365 | 0.01 |
MP2 /6-311++G(d,p) | ||||||
CHF | 475 | 172 | 0.016 | 0.032 | -303 | -0.02 |
CHCl | 593 | 201 | 0.009 | 0.001 | -392 | 0.01 |
CHBr | 613 | 234 | 0.008 | 0.000 | -379 | 0.01 |
Dihalogeno-carbenes |
|
|
|
| ||
|---|---|---|---|---|---|---|
|
|
|
|
|
| |
HF/6-311++G(d,p) | ||||||
CF2 | 115 | 108 | 0.212 | 0.034 | -7 | 0.18 |
CCl2 | 167 | 153 | 0.058 | 0.047 | -14 | 0.01 |
B3LYP /6-311++G(d,p) | ||||||
CF2 | 113 | 168 | 0.216 | 0.027 | 55 | 0.19 |
CCl2 | 138 | 153 | 0.120 | 0.045 | 15 | 0.07 |
MP2 /6-311++G(d,p) | ||||||
CF2 | 125 | 111 | 0.179 | 0.028 | -15 | 0.15 |
CCl2 | 136 | 152 | 0.191 | 0.050 | 16 | 0.14 |
Dihalogeno-carbenes |
|
|
|
| ||
|---|---|---|---|---|---|---|
|
|
|
|
|
| |
HF/6-311++G(d,p) | ||||||
CF2 | 232 | 170 | 0.075 | 0.028 | -62 | 0.05 |
CCl2 | 469 | 236 | 0.009 | 0.001 | -233 | 0.00 |
B3LYP /6-311++G(d,p) | ||||||
CF2 | 237 | 168 | 0.071 | 0.027 | -69 | 0.04 |
CCl2 | 462 | 240 | 0.009 | 0.001 | -222 | 0.01 |
MP2 /6-311++G(d,p) | ||||||
CF2 | 289 | 168 | 0.059 | 0.028 | -121 | 0.03 |
CCl2 | 469 | 235 | 0.009 | 0.001 | -234 | 0.01 |
UV | Ultraviolet |
S0 State | Singlet State |
T1 State | Triplet State |
| [1] |
P. Swings and L. Rosenfeld « Considerations Regarding Interstellar Molecules », Astrophysical Journal, 1937, vol. 86, p. 483-486, DOI 10.1086/143880. [Online]. Available:
https://ui.adsabs.harvard.edu/abs/1937ApJ....86..483S/abstract |
| [2] | J. M. Hollis, P. R. Jewell and F. J. Lovas, «Confirmation of interstellar methylene», Astrophysical Jourmal, 1995, Part 1, vol. 438, P 259-264, |
| [3] | G. Herzberg, «Molecular Spectra and Molecular Structure. III. Electronic Spectra and Electronic Structure of Polyatomic Molecules », D Van Nostrand, Princeton, New-Jersey, 1966, page 336. |
| [4] | M. Krauss, « Triplet state of the carbene radical », Journal of Research of the National Bureau of Standards (J. Res. NBS), 1964, 68A, p. 635. |
| [5] | Balci M. Reaksiyon mekanizmaları. 3th ed. Türkiye Bilimler Akademisi; 2012. 597 p. |
| [6] | Moss RA, Platz MS, Jones, Jr. M., « Reactive Intermediate Chemistry », Wiley-Interscience, 2004. 1067 p. ISBN: 0-471-23324-2. |
| [7] | Nemirowski A, Schreiner PR. « Electronic Stabilization of Ground State Triplet Carbenes », J. Org. Chem. 2007; 72 (25): 9533–9540. |
| [8] | Maroufou Adeyemi Adéoti, « Contribution to the study of the mechanism of action of carbenes on unsaturated bonds »; Thesis, University of Cocody, 1998. |
| [9] | Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., and al. (2009) Gaussian 09, Revision A. 02. Gaussian, Inc. |
| [10] | MJ Frisch, M. Head-Gordon and JA Pople, « Semi-direct algorithms for the MP2 energy and gradient», Chem. Phys. Lett., 1990, 166 281-89. |
| [11] | M. Head-Gordon and T. Head-Gordon, « Analytic MP2 frequencies without fifth-order storage. Theory and application to bifurcated hydrogen bonds in the water hexamer», Chem. Phys. Lett, 1994, 220 (1–2), 122-128. |
| [12] | S. Saebo and J. Almlof, « Avoiding the integral storage bottleneck in LCAO calculations of electron correlation », Chem. Phys. Lett, 1989, 154 (1), 83-89. |
| [13] | P. Hohemberg and W. Kohn, « Inhomogeneous Electron Gas. », Phys. Rev, 1964, 136, B864. |
| [14] | W. Kohn and L. J. Shan, « Self-Consistent Equations Including Exchange and Correlation Effects », Phys. Rev, 1965, 140, A 1133. |
| [15] | Alao Latifatou Laye, « Contribution to the study of carbene properties using Ab initio methods and Density Functional Theory (DFT) from quantum chemistry »; Thesis, University of Félix Houphouët-Boigny, 2019. |
APA Style
Laye, A. L., Sekou, D., Lucie, B. A., Benjamine, A. A., Soleymane, K. (2026). Absorption of Methylene, Hydrohalogenocarbenes and Dihalogenocarbenes in the Ultraviolet Visible Range: Ab Initio and Density Functional Theory (DFT) Approaches. American Journal of Quantum Chemistry and Molecular Spectroscopy, 10(1), 35-41. https://doi.org/10.11648/j.ajqcms.20261001.14
ACS Style
Laye, A. L.; Sekou, D.; Lucie, B. A.; Benjamine, A. A.; Soleymane, K. Absorption of Methylene, Hydrohalogenocarbenes and Dihalogenocarbenes in the Ultraviolet Visible Range: Ab Initio and Density Functional Theory (DFT) Approaches. Am. J. Quantum Chem. Mol. Spectrosc. 2026, 10(1), 35-41. doi: 10.11648/j.ajqcms.20261001.14
AMA Style
Laye AL, Sekou D, Lucie BA, Benjamine AA, Soleymane K. Absorption of Methylene, Hydrohalogenocarbenes and Dihalogenocarbenes in the Ultraviolet Visible Range: Ab Initio and Density Functional Theory (DFT) Approaches. Am J Quantum Chem Mol Spectrosc. 2026;10(1):35-41. doi: 10.11648/j.ajqcms.20261001.14
@article{10.11648/j.ajqcms.20261001.14,
author = {Alao Latifatou Laye and Diomande Sekou and Bede Affoue Lucie and Assoma Amon Benjamine and Kone Soleymane},
title = {Absorption of Methylene, Hydrohalogenocarbenes and Dihalogenocarbenes in the Ultraviolet Visible Range: Ab Initio and Density Functional Theory (DFT) Approaches},
journal = {American Journal of Quantum Chemistry and Molecular Spectroscopy},
volume = {10},
number = {1},
pages = {35-41},
doi = {10.11648/j.ajqcms.20261001.14},
url = {https://doi.org/10.11648/j.ajqcms.20261001.14},
eprint = {https://article.sciencepublishinggroup.com/pdf/10.11648.j.ajqcms.20261001.14},
abstract = {Carbenes are unstable biradical molecules with a longer lifetime in space than on Earth. This theoretical study focused on analyzing the spectroscopic properties of methylene and monohalogenated and dihalogenated derivatives in the ultraviolet-visible range. Structures of these compounds can exist in two electronic states depending on the orbitals containing the non-bonding electrons: singlet S0 and triplet T1. Three theoretical levels HF/6-311++G (d, p), MP2/6-311++G (d, p), and B3LYP/6-311++G (d, p) were used to perform this analysis. Absorption spectra were calculated for the optimized structures (S0 and T1) of methylene, three hydrohalogenocarbenes and two dihalogenocarbenes at each of the chosen theoretical levels. The calculations allowed us to study the behavior of these carbenes in ultraviolet-visible absorption. It was determined that, at maximum absorption, all of these carbenes in the singlet S0 state absorb in the visible range. However, in the T1 triplet state, they absorb in the ultraviolet range. Under maximum oscillation conditions, these carbenes absorb in the ultraviolet regardless of the electronic state of the structure. The calculated absorption wavelengths show that the transition from the ground state to their excited states of the studied carbenes is sometimes accompanied by a bathochromic effect and sometimes by a hypsochromic effect.},
year = {2026}
}
TY - JOUR T1 - Absorption of Methylene, Hydrohalogenocarbenes and Dihalogenocarbenes in the Ultraviolet Visible Range: Ab Initio and Density Functional Theory (DFT) Approaches AU - Alao Latifatou Laye AU - Diomande Sekou AU - Bede Affoue Lucie AU - Assoma Amon Benjamine AU - Kone Soleymane Y1 - 2026/06/10 PY - 2026 N1 - https://doi.org/10.11648/j.ajqcms.20261001.14 DO - 10.11648/j.ajqcms.20261001.14 T2 - American Journal of Quantum Chemistry and Molecular Spectroscopy JF - American Journal of Quantum Chemistry and Molecular Spectroscopy JO - American Journal of Quantum Chemistry and Molecular Spectroscopy SP - 35 EP - 41 PB - Science Publishing Group SN - 2994-7308 UR - https://doi.org/10.11648/j.ajqcms.20261001.14 AB - Carbenes are unstable biradical molecules with a longer lifetime in space than on Earth. This theoretical study focused on analyzing the spectroscopic properties of methylene and monohalogenated and dihalogenated derivatives in the ultraviolet-visible range. Structures of these compounds can exist in two electronic states depending on the orbitals containing the non-bonding electrons: singlet S0 and triplet T1. Three theoretical levels HF/6-311++G (d, p), MP2/6-311++G (d, p), and B3LYP/6-311++G (d, p) were used to perform this analysis. Absorption spectra were calculated for the optimized structures (S0 and T1) of methylene, three hydrohalogenocarbenes and two dihalogenocarbenes at each of the chosen theoretical levels. The calculations allowed us to study the behavior of these carbenes in ultraviolet-visible absorption. It was determined that, at maximum absorption, all of these carbenes in the singlet S0 state absorb in the visible range. However, in the T1 triplet state, they absorb in the ultraviolet range. Under maximum oscillation conditions, these carbenes absorb in the ultraviolet regardless of the electronic state of the structure. The calculated absorption wavelengths show that the transition from the ground state to their excited states of the studied carbenes is sometimes accompanied by a bathochromic effect and sometimes by a hypsochromic effect. VL - 10 IS - 1 ER -
Department of Science, Structures of Matter and Technology (SSMT), University of Felix Houphouët-Boigny, Abidjan, Ivory Coast
Department of Agro-Industrial Sciences and Technologies (AIST), University of San Pedro, San Pedro, Ivory Coast
Department of Science, Structures of Matter and Technology (SSMT), University of Felix Houphouët-Boigny, Abidjan, Ivory Coast
Information