Globally, tuberculosis continues to be a primary cause of death resulting from infectious diseases. The rise of resistant bacterial strains significantly undermines the effectiveness of current treatments. This study examines new sulfonamide derivatives (SULF) that target the Eis enzyme of Mycobacterium tuberculosis. This enzyme plays a direct role in kanamycin resistance. Three-dimensional models of the Eis-SULF complexes were generated from the reference crystal structure (PDB: 5IV0). These complexes were used to construct a test set of thirteen compounds with known experimental activities and an external validation set of four additional compounds. Active conformations were identified using energy-based QSAR modeling. The gas-phase model exhibits an R² coefficient of 0.96. Cross-validation of the gas-phase model yields an R²cv value of 0.95. The standard error of the gas-phase model is 0.28. The model in a solvated environment shows an R² value of 0.97. Cross-validation in a solvated environment yields an R²cv of 0.96. The standard error in a solvated environment is 0.24. A virtual library of sulfonamides was then constructed. The screening was based on Lipinski’s rules and the PH4 model. The PH4 model has an R² value of 0.91. The standard error of the PH4 model is 0.45. Sixty-five compounds show potential for oral bioavailability. The most promising complexes were studied using molecular dynamics. This analysis assesses the stability of ligands in the active site. The MM/GBSA approach was employed to calculate the binding free energies of the complexes. These calculations confirm a high affinity for the Eis enzyme. This integrated approach proposes promising new inhibitors against drug-resistant tuberculosis.
| Published in | American Journal of Chemical and Biochemical Engineering (Volume 10, Issue 1) |
| DOI | 10.11648/j.ajcbe.20261001.11 |
| Page(s) | 1-21 |
| Creative Commons |
This is an Open Access article, distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution and reproduction in any medium or format, provided the original work is properly cited. |
| Copyright |
Copyright © The Author(s), 2026. Published by Science Publishing Group |
Drug-resistant Tuberculosis, Eis Enzyme, Sulfonamide, Molecular Modeling, QSAR, Molecular Dynamics, MM/GBSA
Training set | ||
|---|---|---|
|
|
|
SULF1 | SULF2 | SULF3 |
|
|
|
SULF4 | SULF5 | SULF6 |
|
|
|
SULF7 | SULF8 | SULF9 |
|
|
|
SULF10 ) | SULF11 ) | SULF12 |
| ||
SULF13 | ||
Validation Set | ||
|
|
|
SULF1 | SULF2 | SULF3 |
SULF4 | ||
Training setNew id. | Old id. [17] | Mw [g.mol-1] | ∆∆HMM [kcal.mol-1] | ∆∆Gsol [kcal.mol-1] | ∆∆TSvib [kcal.mol-1] | ∆∆Gcom [kcal.mol-1] |
| |
|---|---|---|---|---|---|---|---|---|
SULF1 | 46 | 0,24 | 381 | 0.00 | 0.00 | 0.00 | 0.00 | 9.62 |
SULF2 | 42 | 27 | 359 | 12.45 | -0.97 | 1.74 | 9.73 | 7.57 |
SULF3 | 38 | 56 | 410 | 15.89 | -3.32 | 1.28 | 11.29 | 7.25 |
SULF4 | 29 | 80 | 410 | 16.97 | -3.43 | 0.84 | 12.70 | 7.10 |
SULF5 | 35 | 100 | 366 | 17.25 | -3.55 | 1.21 | 12.49 | 7.00 |
SULF6 | 43 | 230 | 373 | 21.61 | 1.48 | 4.58 | 18.52 | 6.64 |
SULF7 | 44 | 700 | 387 | 22.70 | 0.61 | 6.47 | 16.84 | 6.15 |
SULF8 | 37 | 3000 | 349 | 23.55 | 1.87 | 3.24 | 22.18 | 5.52 |
SULF9 | 39 | 5800 | 361 | 25.11 | -0.12 | 2.92 | 22.07 | 5.24 |
SULF10 | 31 | 5800 | 331 | 24.69 | 2.30 | 4.39 | 22.60 | 5.24 |
SULF11 | 30 | 6200 | 396 | 25.04 | -3.95 | 0.25 | 20.84 | 5.21 |
SULF12 | 34 | 10600 | 359 | 26.06 | 2.01 | 3.81 | 24.26 | 4.97 |
SULF13 | 45 | 27000 | 387 | 30.59 | -0.37 | 4.54 | 25.68 | 4.57 |
Validation Set | MW [g.mol-1] | ∆∆HMM [kcal.mol-1] | ∆∆Gsol [kcal.mol-1] | ∆∆TSvib [kcal.mol-1] | ∆∆Gcom [kcal.mol-1] | / p | ||||
|---|---|---|---|---|---|---|---|---|---|---|
New id. | Old id. [17] | |||||||||
SULF1 | 33 | 250 | 6.60 | 373 | 16.89 | 2.69 | 5.13 | 14.45 | 6.77 | 1.03 |
SULF2 | 47 | 250 | 6.52 | 411 | 17.09 | 1.27 | 0.75 | 17.61 | 6.17 | 0.95 |
SULF3 | 41 | 370 | 6.43 | 345 | 20.25 | 2.10 | 3.06 | 19.29 | 5.85 | 0.91 |
SULF4 | 40 | 7400 | 5.13 | 391 | 23.08 | 0.16 | 1.58 | 21.65 | 5.41 | 1.05 |
Statistical Data of Linear Regression | (A) | (B) |
|---|---|---|
| ||
Number of compounds n | 13 | 13 |
Squared correlation coefficient of regression R2 | 0.96 | 0.97 |
LOO cross-validated squared correlation coefficient | 0.95 | 0.96 |
Standard error of regression | 0.28 | 0.24 |
Statistical significance of regression, Fisher F-test | 270.55 | 371.64 |
Level of statistical significance α | > 95% | > 95% |
Range of activities [17] | 0.24 - 27000 | |
Hypothesis | RSMD a | R2 b | Total Cost c | Costs Difference d | Closest Random e | Features f |
|---|---|---|---|---|---|---|
Hypo 1 | 6.085 | 0.95 | 307.1 | 2250.1 | 354.17 | HBA, HBD, HYD, HYD, HYD |
Hypo 2 | 6.260 | 0.94 | 323.2 | 2234.0 | 414.84 | HBA, HBD, HYD, HYD, HYD |
Hypo 3 | 8.067 | 0.91 | 482.9 | 2074.3 | 468.24 | HBA, HBD, HYD, HYD |
Hypo 4 | 8.332 | 0.90 | 497.2 | 2060.0 | 495.72 | HBA, HBD, HYD, HYD, HYD |
Hypo 5 | 8.458 | 0.90 | 505.1 | 2052.1 | 618.78 | HBA, HBD, HYD, HYD, HYD |
Hypo 6 | 8.514 | 0.90 | 522.0 | 2035.2 | 644.83 | HBA, HBD, HYD, HYD |
Hypo 7 | 8.733 | 0.89 | 548,0 | 2009.2 | 647.29 | HBD, HYD, HYD, HYD |
Hypo 8 | 8,723 | 0,89 | 556.8 | 2000.4 | 661.16 | HBA, HYD, HYD, HYD |
Hypo 9 | 8.797 | 0,89 | 559.5 | 1997.7 | 661.79 | HBA, HBD, HYD, HYD, HYD |
Hypo 10 | 9.105 | 0.88 | 582.1 | 1975.1 | 674.17 | HBA, HBD, HYD, HYD, HYD |
Fixed Cost | 0 | 0 | 29.9 | |||
Null Cost | 19.763 | 0 | 2557.2 |
Designed analogs | Substituents | Mw [g.mol-1] | ∆∆HMM [kca.mol-1] | ∆∆Gsol [kca.mol-1] | ∆∆TSvib [kca.mol-1] | ∆∆Gcom [kca.mol-1] | ||
|---|---|---|---|---|---|---|---|---|
R1 | R2 | |||||||
SULF1 | CH3 | naphtalen-2-yl | 381 | 0.00 | 0.00 | 0.00 | 0.00 | 9.62* |
1-239 | H | 3-bromonaphtalen-2-yl | 446 | 13.23 | 1.05 | 0.90 | 13.38 | 6.67 |
1-444 | H | 6-bromo-2H-chromen-7-yl | 450 | 1.32 | 0.64 | 2.30 | -0.34 | 9.56 |
1-450 | H | 6-hydroxy-2H-chromen-7-yl | 401 | -0.11 | 0.83 | 3.12 | -2.39 | 9.95 |
2-84 | F | 2-chloro-5-methoxyphenyl | 400 | 3.09 | 0.97 | 4.79 | -0.73 | 9.64 |
2-113 | F | 3-ethoxyphenyl | 379 | 3.36 | 0.22 | 3.43 | 0.15 | 9.47 |
2-234 | F | 4-methoxynaphthalen-2-yl | 415 | 6.07 | 2.51 | 1.97 | 6.62 | 8.25 |
2-240 | F | 1-bromonaphthalen-2-yl | 464 | 6.86 | -0.43 | 1.33 | 5.10 | 8.54 |
2-388 | F | 7-bromo-1H-indol-6-yl | 453 | 4.63 | 0.55 | 0.17 | 5.01 | 8.55 |
3-40 | Cl | 5-chloro-2-methylphenyl | 400 | -1.89 | -0.50 | -0.16 | -2.55 | 9.98 |
3-51 | Cl | 3-chloro-2-(trifluoromethyl)phenyl | 454 | 9.26 | 0.19 | -4.19 | 13.65 | 6.92 |
3-66 | Cl | 2.5-dimethylphenyl | 380 | 9.92 | -0.04 | 0.98 | 8.90 | 7.82 |
3-148 | Cl | 2- [4-(propan-2-yl)phenyl] ethyl | 422 | 7.35 | -0.30 | 4.14 | 2.90 | 8.95 |
3-311 | Cl | 7-fluoro-1H-inden-5-yl | 408 | 2.78 | 1.08 | -0.76 | 4.62 | 8.63 |
3-420 | Cl | 1-benzofuran-6-yl | 392 | 1.15 | 0.15 | 1.41 | -0.12 | 9.52 |
3-432 | Cl | 6-fluoro-2H-chromen-7-yl | 424 | -2.15 | 0.38 | 1.42 | -3.18 | 10.10 |
4-257 | Br | 4.6-dimethylnaphthalen-2-yl | 474 | 0.40 | 0.42 | -0.16 | 0.98 | 9.32 |
4-378 | Br | 1H-indol-5-yl | 435 | 0.14 | 2.59 | 0.78 | 1.95 | 9.13 |
4-424 | Br | 4-methyl-2H-chromen-7-yl | 464 | -1.52 | 0.03 | -0.14 | -1.35 | 9.76 |
4-434 | Br | 7-chloro-2H-chromen-2-yl | 485 | 3.31 | 0.13 | -1.03 | 4.47 | 8.66 |
5-81 | I | 2-chlorophenyl | 478 | 3.61 | 0.23 | -1.35 | 5.20 | 8.52 |
5-223 | I | 6-methoxypyrimidin-4-yl | 475 | 8.45 | -1.68 | -0.25 | 7.02 | 8.17 |
5-399 | I | 6-methyl-2-benzofuran-5-yl | 497 | 6.30 | 0.48 | 0.28 | 6.50 | 8.27 |
6-234 | CH3 | 4-methoxynaphthalen-2-yl | 411 | 2.60 | 1.99 | -0.78 | 5.37 | 8.49 |
6-269 | CH3 | 1,4-dimethylnaphthalen-2-yl | 409 | -2,50 | -1,23 | 0,50 | -4,23 | 10,30 |
6-444 | CH3 | 6-bromo-8aH-chromen-7-yl | 464 | 1.42 | 0.07 | 0.35 | 1.14 | 9.29 |
6-496 | CH3 | thianthren-2-yl | 470 | -4.91 | 4.99 | -2.68 | 2.77 | 8.98 |
6-522 | CH3 | 1.4.5.8-tetramethylnaphthalen-2-yl | 438 | -1.54 | 0.05 | 1.82 | -3.31 | 10.13 |
7-109 | C2H5 | methyl 2-aminobenzoate | 403 | 14.56 | -0.32 | -1.25 | 15.49 | 6.57 |
7-127 | C2H5 | 2-(3-methylphenyl)ethyl | 371 | 5.94 | 0.07 | 2.54 | 3.47 | 8.84 |
7-141 | C2H5 | 2-(2-ethylphenyl)ethyl | 385 | 11.56 | -0.03 | 5.51 | 6.01 | 8.36 |
7-239 | C2H5 | 3-bromonaphtalen-2-yl | 474 | 8.48 | -0.38 | 1.22 | 6.88 | 8.20 |
7-267 | C2H5 | 5.8-dimethylnaphthalen-2-yl | 423 | -1.12 | -3.24 | 5.97 | -10.33 | 11.45 |
7-269 | C2H5 | 1.4-dimethylnaphthalen-2-yl | 423 | 3.84 | -2.47 | 7.48 | -6.11 | 10.65 |
7-270 | C2H5 | 1.3-dimethylnaphthalen-2-yl | 423 | 7.85 | -1.98 | 6.21 | -0.34 | 9.57 |
7-271 | C2H5 | naphthalen-1-yl | 395 | 1.29 | 0.58 | 1.99 | -0.11 | 9.52 |
7-272 | C2H5 | 2-methylnaphthalen-1-yl | 409 | 4.45 | -0.16 | 2.75 | 1.54 | 9.21 |
7-273 | C2H5 | 3-methylnaphthalen-1-yl | 409 | 1.92 | -0.30 | 4.03 | -2.42 | 9.96 |
7-274 | C2H5 | 4-methylnaphthalen-1-yl | 409 | 0.40 | 0.88 | 2.03 | -0.75 | 9.64 |
7-275 | C2H5 | 5-methylnaphthalen-1-yl | 409 | 0.67 | 1.33 | -0.24 | 2.24 | 9.08 |
7-276 | C2H5 | 6-methylnaphthalen-1-yl | 409 | 1.27 | 0.58 | 2.95 | -1.09 | 9.71 |
7-277 | C2H5 | 7-methylnaphthalen-1-yl | 409 | 0.73 | 1.14 | -0.29 | 2.16 | 9.09 |
7-278 | C2H5 | 8-methylnaphthalen-1-yl | 409 | 5.39 | -2.42 | 2.95 | 0.03 | 9.50 |
7-279 | C2H5 | 2-chloronaphthalen-1-yl | 430 | -0.58 | -0.62 | 2.00 | -3.20 | 10.10 |
7-388 | C2H5 | 7-bromo-1H-indol-6-yl | 463 | 4.29 | 0.55 | -0.93 | 5.76 | 8.41 |
7-522 | C2H5 | 1.4.5.8-tetramethylnaphthalen-2-yl | 452 | -0.32 | -2.61 | 3.00 | -5.93 | 10.62 |
7-524 | C2H5 | 1.4.5.6.8-pentamethylnaphthalen-2-yl | 466 | 1.40 | -3.53 | 6.69 | -8.83 | 11.17 |
7-525 | C2H5 | 1.4.5.7.8-pentamethylnaphthalen-2-yl | 466 | 1.80 | -4.04 | 8.85 | -11.09 | 11.60 |
7-526 | C2H5 | 4.5.6.8-tetramethylnaphthalen-2-yl | 452 | -1.01 | -5.40 | 4.43 | -10.85 | 11.55 |
8-20 | isopropyl | 3-tert-butylphenyl | 416 | 10.60 | -4.43 | 7.00 | -0.83 | 9.66 |
8-30 | isopropyl | 4-methoxyphenyl | 389 | 10.18 | -1.90 | 1.90 | 6.38 | 8.29 |
8-315 | isopropyl | 1H-inden-6-yl | 397 | 7.67 | -0.60 | 0.85 | 6.22 | 8.32 |
8-331 | isopropyl | 2-methoxy-3H-inden-5-yl | 427 | -1.52 | 2.15 | 3.55 | -2.91 | 10.05 |
8-332 | isopropyl | 3-methoxy-1H-inden-5-yl | 427 | 2.96 | -0.17 | 2.42 | 0.36 | 9.43 |
8-333 | isopropyl | 1-bromo-1H-inden-5-yl | 476 | 1.51 | -0.71 | 1.85 | -1.05 | 9.70 |
8-334 | isopropyl | 7-bromo-1H-inden-5-yl | 476 | 0.72 | 0.13 | 3.10 | -2.25 | 9.93 |
8-335 | isopropyl | 6-bromo-1H-inden-5-yl | 476 | 2.05 | 0.78 | 2.01 | 0.81 | 9.35 |
8-336 | isopropyl | 6-methoxy-1H-inden-5-yl | 427 | 1.96 | -0.04 | 2.75 | -0.83 | 9.66 |
8-337 | isopropyl | 7-methoxy-1H-inden-5-yl | 427 | 0.77 | 0.29 | -3.53 | 4.59 | 8.63 |
8-338 | isopropyl | 1-methoxy-1H-inden-5-yl | 427 | 1.23 | -0.27 | -1.48 | 2.43 | 9.04 |
8-339 | isopropyl | 2-methoxy-1H-inden-5-yl | 427 | -0.71 | -0.07 | -0.67 | -0.12 | 9.52 |
8-353 | isopropyl | 4-methylpentalen-2-yl | 397 | 7.87 | -0.31 | -0.43 | 7.99 | 7.99 |
8-361 | isopropyl | 1-hydroxyindol-6-yl | 414 | 5.63 | 0.81 | 0.86 | 5.58 | 8.45 |
8-435 | isopropyl | 7-chloro-2H-chromen-3-yl | 448 | 15.14 | -1.34 | 3.91 | 9.88 | 7.63 |
8-468 | isopropyl | 5-chloro-2H-chromen-7-yl | 448 | 5.44 | 0.91 | -2.80 | 9.15 | 7.77 |
9-143 | tert-butyl | 2-(2-tert-butylphenyl)ethyl | 458 | 40.60 | 3.77 | 8.68 | 35.70 | 2.75 |
SULFx | HBdon | HBacc | logPo/w | logSwat | logKHSA | BIPcaco [nm.s-1] | #meta | [nM] | HOA | %HOA |
|---|---|---|---|---|---|---|---|---|---|---|
7-525 | 2 | 9.5 | 2.4 | -5.5 | 0.23 | 92 | 5 |
| 3 | 76 |
7-526 | 2 | 9.5 | 2.7 | -5.8 | 0.41 | 93 | 7 |
| 2 | 78 |
7-269 | 2 | 9.5 | 1.9 | -4.7 | -0.01 | 95 | 3 |
| 3 | 73 |
6-269 | 2 | 9.5 | 1.6 | -4.6 | -0.1 | 90 | 3 |
| 3 | 71 |
Rifampicine | 3 | 4.5 | -0.7 | 0 | -0.8 | 267.5 | 2 | - | 2 | 67 |
Isoniazid | 2 | 5 | -0.6 | -0.5 | -0.8 | 298.4 | 4 | - | 2 | 67 |
Ethambutol | 4 | 6.4 | -0.2 | 0.6 | -0.8 | 107.8 | 4 | - | 2 | 62 |
Pyrazinamide | 6 | 20.3 * | 3.0 | -3.1 | -0.3 | 38.2 | 11 * | - | 1 | 34 |
Gatifloxacin | 1 | 6.8 | 0.5 | -4.0 | 0 | 17.0 | 1 | - | 2 | 52 |
Moxifloxacin | 1 | 6.8 | 1.0 | -4.7 | 0.2 | 20.9 | 1 | - | 2 | 56 |
Rifapentine | 6 | 20.9* | 3.6 | -2.2 | -0.2 | 224.40 | 13 | - | 1 | 51 |
Bedaquiline | 1 | 3.8 | 7.6* | -6.9 | 1.7 | 1562.2 | 5 | - | 1 | 100 |
Delamanid | 0 | 6.0 | 5.8 | -7.6 | 1.0 | 590.9 | 2 | - | 1 | 85 |
Linezolid | 1 | 8.7 | 0.6 | -2.0 | -0.7 | 507.0 | 2 | - | 3 | 79 |
Sutezolid | 1 | 7.5 | 1.3 | -3.4 | -0.4 | 449.3 | 0 | - | 3 | 82 |
Ofloxacin | 0 | 7.3 | -0.4 | -2.8 | -0.5 | 25.9 | 1 | - | 2 | 50 |
Amikacin | 17* | 26.9* | -7.9* | -0.2 | -2.1 | 0 | 14* | - | 1 | 0 |
Kanamycin | 15* | 227* | -6.7* | 2.0 | -1.4 | 0 | 12* | - | 1 | 0 |
Imipenem | 3 | 7.2 | 1.0 | -1.8 | -0.7 | 35.0 | 3 | - | 3 | 61 |
Amoxicillin | 4,25 | 8.0 | -2.5 | -0.8 | -1.1 | 1.0 | 5 | - | 1 | 12 |
Clavulanate | 2 | 6.5 | -0.8 | 0.3 | -1.3 | 13.3 | 2 | - | 2 | 42 |
Inhibitors | Range | Standard deviation | MM-GBSA | (pM) | ||
|---|---|---|---|---|---|---|
SULF1 | -89.72 to -54.90 | 6.03 | -70.4677 | 0.00 | 0.00 | 240 |
SULF7-525 | -105.03 to -73.67 | 5.87 | -88.0910 | -17.6233 | -11.09 | 2.51 |
SULF7-526 | -110.87 to -57.10 | 8.78 | -85.7695 | -15.3018 | -10.85 | 2.81 |
SULF7-269 | -101.21 to -56.17 | 6.26 | -81.6070 | -11.1393 | -6.11 | 22.4 |
SULF6-269 | -104.06 to -45.58 | 8.44 | -74.7527 | -4.285 | -4.23 | 50.1 |
ADME | Absorption. Distribution. Metabolism. and Excretion |
2D | Two-Dimensional |
3D | Three-Dimensional |
QSAR | Quantitative Structure-Activity Relationships |
GFE | Gibbs Free Energy |
VCL | Virtual Combinatorial Library |
DHODH | Dihydroorotate Dehydrogenase |
SULF | Sulfonamide |
Eis | Enhance Intracellular Survival |
| Experimentally Half-Maximal Inhibitory Concentration |
PH4 | pharmacophore |
WHO | World Health Organization |
Mtb | Mycobacterium tuberculosis |
VL | Virtual Library |
TS | Test Set |
VS | Validation Set |
E-I | Enzyme-Inhibitor |
Eint | Enzyme–Inhibitor Interaction Energy per Residue |
| Relative Complexation GFE |
| Predicted Half-Maximal Inhibitory Concentration |
MD | Molecular Dynamics |
MM | Molecular Mechanics |
GBSA | Generalized Born Surface Area |
| [1] | World Health Organization. Global Tuberculosis Report 2025. Geneva: WHO; 2025. |
| [2] | Dheda K, Gumbo T, Maartens G, Dooley KE, McNerney R, Murray M, et al. The epidemiology, pathogenesis, transmission, diagnosis, and management of multidrug-resistant, extensively drug-resistant, and incurable tuberculosis. Lancet Respir Med. 2017; 5(4): 291-360. |
| [3] | Gandhi NR, Nunn P, Dheda K, Schaaf HS, Zignol M, Van Soolingen D, et al. Multidrug-resistant and extensively drug-resistant tuberculosis: a threat to global control of tuberculosis. Lancet. 2010; 375(9728): 1830-1843. |
| [4] | Chen W, Biswas T, Porter VR, Tsodikov OV, Garneau-Tsodikova S. Unusual regioversatility of acetyltransferase Eis, a cause of drug resistance in XDR-TB. Proc Natl Acad Sci U S A. 2011; 108(24): 9815-9820. |
| [5] | Willby MJ, Green KD, Gajadeera CS, Hou C, Tsodikov OV, Posey JE, Garneau-Tsodikova S. Potent Inhibitors of Acetyltransferase Eis Overcome Kanamycin Resistance in Mycobacterium tuberculosis. ACS Chem Biol. 2016; 11(6): 1639-1646. |
| [6] | Zaunbrecher MA, Sikes RD Jr, Metchock B, Shinnick TM, Posey JE. Overexpression of the chromosomally encoded aminoglycoside acetyltransferase Eis confers kanamycin resistance in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2009; 106(47): 20004-20009. |
| [7] | Shin DM, Jeon BY, Lee HM, Jin HS, Yuk JM, Song CH, et al. Mycobacterium tuberculosis Eis regulates autophagy, inflammation, and cell death through redox-dependent signaling. PLoS Pathog. 2010; 6(12): e1001230. |
| [8] | Jennings BC, Labby KJ, Green KD, Garneau-Tsodikova S. Redesign of substrate specificity and identification of the aminoglycoside binding residues of Eis from Mycobacterium tuberculosis. Biochemistry. 2013; 52: 5125-5132. |
| [9] | Punetha A, Ngo HX, Holbrook SYL, Green KD, Willby MJ, Bonnett SA, et al. Structure-Guided Optimization of Inhibitors of Acetyltransferase Eis from Mycobacterium tuberculosis. ACS Chem Biol. 2020; 15(6): 1581-1594. |
| [10] | Houghton JL, Biswas T, Chen W, Tsodikov OV, Garneau-Tsodikova S. Chemical and structural insights into the regioversatility of the aminoglycoside acetyltransferase Eis. ChemBioChem. 2013; 14: 2127-2135. |
| [11] | Ovung A, Bhattacharyya J. Sulfonamide drugs: structure, antibacterial property, toxicity, and biophysical interactions. Biophys Rev. 2021; 13(2): 259-272. |
| [12] | Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov. 2004; 3: 935-949. |
| [13] | Roy K, Kar S, Das RN. A Primer on QSAR/QSPR Modeling. Springer; 2015. |
| [14] | Hollingsworth SA, Dror RO. Molecular dynamics simulation for all. Neuron. 2018; 99(6): 1129-1143. |
| [15] | Hou T, Wang J, Li Y, Wang W. Assessing the performance of MM/PBSA and MM/GBSA methods. J Chem Inf Model. 2011; 51(1): 69-82. |
| [16] | Conradie F, Diacon AH, Ngubane N, et al. Treatment of highly drug-resistant pulmonary tuberculosis. N Engl J Med. 2020; 382: 893-902. |
| [17] | Garzan A, Willby MJ, Green KD, et al. Sulfonamide-based inhibitors of Eis abolish kanamycin resistance in Mycobacterium tuberculosis. ACS Infect Dis. 2016; 2(6): 423-430. |
| [18] | OECD. Guidance Document on the Validation of (Quantitative) Structure-Activity Relationship [(Q)SAR] Models; OECD Series on Testing and Assessment, No. 69; OECD Publishing: Paris, France, 2014. |
| [19] | Insight-II and Discover Molecular Modeling and Simulation Package, version 2005; Accelrys: San Diego, CA, USA, 2005. |
| [20] | Discovery Studio molecular modeling and simulation program, Version 2.5, Accelrys, Inc., San Diego, CA, 2009. |
| [21] | Niaré A, Alex AYZ, Bernard DAM, Marius KS, Stéphane DS, Moise KA, Guy-Richard KM, Fagnidi YKH, Doh S. Study by molecular docking of the interactions between dihydroorotate dehydrogenase and a series of inhibitors of pyrrole derivatives for the treatment of malaria. Asian Journal of Chemical Sciences. 2025; 15(1): 92-110. |
| [22] | Soro I, N'Guessan H, Abou A, N'Guessan RK, Megnassan E. Conformational study of molecules in a biological environment, design of inhibitors of human aminopeptidase M1 implicated in cancer therapy. Universal J Pharm Res. 2023; 8(5): 71-86. |
| [23] | Kouadja RJ, Mousse LM, Kouman KC, Nsangou M, Megnassan E. Computer-assisted design of benzoisoxazol derivatives inhibitors of bromodomain-containing protein 4 (BRD4) with favorable pharmacokinetic profile. Int J Pharm Sci Drug Res. 2023; 15(5): 647-664. |
| [24] | Niaré A, N'Guessan H, Dali B, Megnassan E. Computer-assisted design of hydroxamic acid derivatives inhibitors of M1 metallo aminopeptidase of Plasmodium falciparum with favorable pharmacokinetic profile. J Pharm Res Int. 2022; 34(60): 21-44. |
| [25] | Maple JR, Hwang M, Stockfisch TP, Dinur U, Waldman M, Ewig CS, Hagler AT. Derivation of class II force fields. I. Methodology and quantum force field for the alkyl functional group and alkane molecules. J Comput Chem. 1994; 15: 162-182. |
| [26] | Bieri C, Esmel A, Keita M, Owono LCO, Dali B, Megnassan E, Miertus S, Frecer V. Structure-Based Design and Pharmacophore-Based Virtual Screening of Combinatorial Library of Triclosan Analogs Active against Enoyl-Acyl Carrier Protein Reductase of Plasmodium falciparum with Favourable ADME Profiles. Int J Mol Sci. 2023; 24: 6916. |
| [27] | Gilson MK, Barry HH. The inclusion of electrostatic hydration energies in molecular mechanics calculations. J Comput Aided Mol Des. 1991; 5(1): 5-20. |
| [28] | Miertuš S, Scrocco E, Tomasi J. Electrostatic interaction of a solute with a continuum. A direct utilization of ab initio molecular potentials for the prevision of solvent effects. Chem Phys. 1981; 55: 117-129. |
| [29] | Niaré A, N'Guessan A-B, Djako BA, Dembélé GS, Koné MG-R, Yéo Y. QSAR, pharmacophore, and molecular docking studies for the design of novel arylamide-derived inhibitors of Mycobacterium tuberculosis. Chemical Science International Journal. 2024; 33(6): 212-224. |
| [30] | Fischer S, Smith JC, Verma CS. Dissecting the vibrational entropy change on protein/ligand binding: Burial of a water molecule in bovine pancreatic trypsin inhibitor. J Phys Chem B. 2001; 105: 8050-8055. |
| [31] | Fofana I, Dali B, Koné M, Sujova K, Megnassan E, Miertus S, Frecer V. In Silico Optimization of Inhibitors of the 3-Chymotrypsin-like Protease of SARS-CoV-2. Life. 2025; 16(1): 6. |
| [32] | Wienen-Schmidt B, Jonker HR, Wulsdorf T, Gerber HD, Saxena K, Kudlinzki D, Klebe G. Paradoxically, most flexible ligand binds most entropy-favored: intriguing impact of ligand flexibility and solvation on drug-kinase binding. J Med Chem. 2018; 61(14): 5922-5933. |
| [33] | QikProp; Version 3.7, Release 14, X Schrödinger; LLC: New York, NY, USA, 2014. |
| [34] | Desmond Molecular Dynamics System. Release 2021-2; Schrödinger LLC: New York, NY, USA, 2021. |
| [35] | Frecer V, Miertus S. Antiviral agents against COVID-19: Structure-based design of specific peptidomimetic inhibitors of SARS-CoV-2 main protease. RSC Adv. 2020; 10: 40244-40263. |
| [36] | Wang X, Xu Y, Zheng H, Yu K. An Extendible, Graph-Neural-Network-Based Approach for Accurate Force Field Development of Large Flexible Organic Molecules. arXiv preprint arXiv: 2106.00927. 2021. |
| [37] | Herbert JM. Dielectric continuum methods for quantum chemistry. WIREs Comput Mol Sci. 2022; 12(1): e1519. |
| [38] | Kansal N, Silakari O, Ravikumar M. Three dimensional pharmacophore modelling for c-Kit receptor tyrosine kinase inhibitors. Eur J Med Chem. 2010; 45(1): 393-404. |
| [39] | Barber C, Heghes C, Johnston L. A framework to support the application of the OECD guidance documents on (Q) SAR model validation and prediction assessment for regulatory decisions. Comput Toxicol. 2024; 30: 100305. |
| [40] | N'Guessan H, Soro I, Keita M, Megnassan E. Design and In silico Screening of Combinatorial Library of New Herbicidal Analogs of Cycloalka [d] quinazoline2, 4dione-Benzoxazinones Inhibiting Protoporphyrinogen IX Oxidase. J Pharm Res Int. 2022; 34(56): 42-61. |
| [41] | Keita M, Yaya Y, Yvon BMB, Esmel AE, Dali B, N'Guessan H. Molecular and Thermodynamic Modeling of the Protein-Ligand Interaction. Application to Computer-Assisted Design of Anti-Competitive Inhibitors of Human Histone Deacetylase. 2021; 2: 606-630. |
| [42] | Adama N, N'Guessan H, Dali B, Megnassan E. Computer-assisted design of hydroxamic acid derivatives inhibitors of M1 Metallo Aminopeptidase of Plasmodium falciparum with favorable pharmacokinetic profile. J Pharm Res Int. 2022; 21-44. |
| [43] | Revillo Imbernon J, Jacquemard C, Bret G, Marcou G, Kellenberger E. Comprehensive analysis of commercial fragment libraries. RSC Med Chem. 2022; 13(3): 300-310. |
| [44] | Davis JM, Smith LR. Advances in computational approaches for estimating passive permeability in drug discovery. Membranes. 2023; 13(11): 851. |
| [45] | QikProp 6.5. Release 139; Schrödinger LLC.: New York, NY, USA, 2019. |
APA Style
Ibrahim, B., Justin, K. R., Mathias, M. L., Issouf, S., Adama, N., et al. (2026). Molecular Modelling Applied to the Computer-aided Design of Inhibitors of Mycobacterium Tuberculosis Aminoglycoside Acetyltransferase (Eis) to Combat Kanamycin Resistance. American Journal of Chemical and Biochemical Engineering, 10(1), 1-21. https://doi.org/10.11648/j.ajcbe.20261001.11
ACS Style
Ibrahim, B.; Justin, K. R.; Mathias, M. L.; Issouf, S.; Adama, N., et al. Molecular Modelling Applied to the Computer-aided Design of Inhibitors of Mycobacterium Tuberculosis Aminoglycoside Acetyltransferase (Eis) to Combat Kanamycin Resistance. Am. J. Chem. Biochem. Eng. 2026, 10(1), 1-21. doi: 10.11648/j.ajcbe.20261001.11
AMA Style
Ibrahim B, Justin KR, Mathias ML, Issouf S, Adama N, et al. Molecular Modelling Applied to the Computer-aided Design of Inhibitors of Mycobacterium Tuberculosis Aminoglycoside Acetyltransferase (Eis) to Combat Kanamycin Resistance. Am J Chem Biochem Eng. 2026;10(1):1-21. doi: 10.11648/j.ajcbe.20261001.11
@article{10.11648/j.ajcbe.20261001.11,
author = {Bamba Ibrahim and Kouadja Rika Justin and Mousse Logbo Mathias and Soro Issouf and Niare Adama and Megnassan Eugene},
title = {Molecular Modelling Applied to the Computer-aided Design of Inhibitors of Mycobacterium Tuberculosis Aminoglycoside Acetyltransferase (Eis) to Combat Kanamycin Resistance},
journal = {American Journal of Chemical and Biochemical Engineering},
volume = {10},
number = {1},
pages = {1-21},
doi = {10.11648/j.ajcbe.20261001.11},
url = {https://doi.org/10.11648/j.ajcbe.20261001.11},
eprint = {https://article.sciencepublishinggroup.com/pdf/10.11648.j.ajcbe.20261001.11},
abstract = {Globally, tuberculosis continues to be a primary cause of death resulting from infectious diseases. The rise of resistant bacterial strains significantly undermines the effectiveness of current treatments. This study examines new sulfonamide derivatives (SULF) that target the Eis enzyme of Mycobacterium tuberculosis. This enzyme plays a direct role in kanamycin resistance. Three-dimensional models of the Eis-SULF complexes were generated from the reference crystal structure (PDB: 5IV0). These complexes were used to construct a test set of thirteen compounds with known experimental activities and an external validation set of four additional compounds. Active conformations were identified using energy-based QSAR modeling. The gas-phase model exhibits an R² coefficient of 0.96. Cross-validation of the gas-phase model yields an R²cv value of 0.95. The standard error of the gas-phase model is 0.28. The model in a solvated environment shows an R² value of 0.97. Cross-validation in a solvated environment yields an R²cv of 0.96. The standard error in a solvated environment is 0.24. A virtual library of sulfonamides was then constructed. The screening was based on Lipinski’s rules and the PH4 model. The PH4 model has an R² value of 0.91. The standard error of the PH4 model is 0.45. Sixty-five compounds show potential for oral bioavailability. The most promising complexes were studied using molecular dynamics. This analysis assesses the stability of ligands in the active site. The MM/GBSA approach was employed to calculate the binding free energies of the complexes. These calculations confirm a high affinity for the Eis enzyme. This integrated approach proposes promising new inhibitors against drug-resistant tuberculosis.},
year = {2026}
}
TY - JOUR T1 - Molecular Modelling Applied to the Computer-aided Design of Inhibitors of Mycobacterium Tuberculosis Aminoglycoside Acetyltransferase (Eis) to Combat Kanamycin Resistance AU - Bamba Ibrahim AU - Kouadja Rika Justin AU - Mousse Logbo Mathias AU - Soro Issouf AU - Niare Adama AU - Megnassan Eugene Y1 - 2026/05/30 PY - 2026 N1 - https://doi.org/10.11648/j.ajcbe.20261001.11 DO - 10.11648/j.ajcbe.20261001.11 T2 - American Journal of Chemical and Biochemical Engineering JF - American Journal of Chemical and Biochemical Engineering JO - American Journal of Chemical and Biochemical Engineering SP - 1 EP - 21 PB - Science Publishing Group SN - 2639-9989 UR - https://doi.org/10.11648/j.ajcbe.20261001.11 AB - Globally, tuberculosis continues to be a primary cause of death resulting from infectious diseases. The rise of resistant bacterial strains significantly undermines the effectiveness of current treatments. This study examines new sulfonamide derivatives (SULF) that target the Eis enzyme of Mycobacterium tuberculosis. This enzyme plays a direct role in kanamycin resistance. Three-dimensional models of the Eis-SULF complexes were generated from the reference crystal structure (PDB: 5IV0). These complexes were used to construct a test set of thirteen compounds with known experimental activities and an external validation set of four additional compounds. Active conformations were identified using energy-based QSAR modeling. The gas-phase model exhibits an R² coefficient of 0.96. Cross-validation of the gas-phase model yields an R²cv value of 0.95. The standard error of the gas-phase model is 0.28. The model in a solvated environment shows an R² value of 0.97. Cross-validation in a solvated environment yields an R²cv of 0.96. The standard error in a solvated environment is 0.24. A virtual library of sulfonamides was then constructed. The screening was based on Lipinski’s rules and the PH4 model. The PH4 model has an R² value of 0.91. The standard error of the PH4 model is 0.45. Sixty-five compounds show potential for oral bioavailability. The most promising complexes were studied using molecular dynamics. This analysis assesses the stability of ligands in the active site. The MM/GBSA approach was employed to calculate the binding free energies of the complexes. These calculations confirm a high affinity for the Eis enzyme. This integrated approach proposes promising new inhibitors against drug-resistant tuberculosis. VL - 10 IS - 1 ER -
Laboratory of Fundamental and Applied Physics, Nangui Abrogoua University, Abidjan, Côte d’Ivoire
Figure 1. Structure of compound 46.
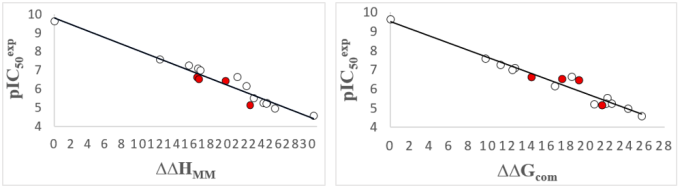
Figure 2. (Left) Linear regression analysis comparing the empirical values against the computed enthalpic term (∆∆HMM [kcal.mol-1]), (Right) An equivalent graphical representation illustrating the relationship with the total complexation Gibbs free energy (∆∆Gcom [kcal.mol-1]) for the training molecules [17], Red markers identify the validation set compounds.
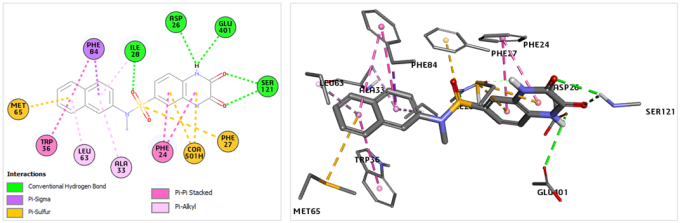
Figure 3. (Left) Two-dimensional representation mapping the binding interactions of the highly active SULF1 molecule within the Eis catalytic pocket. (Right) Three-dimensional visualization detailing the same enzyme-ligand spatial arrangement.
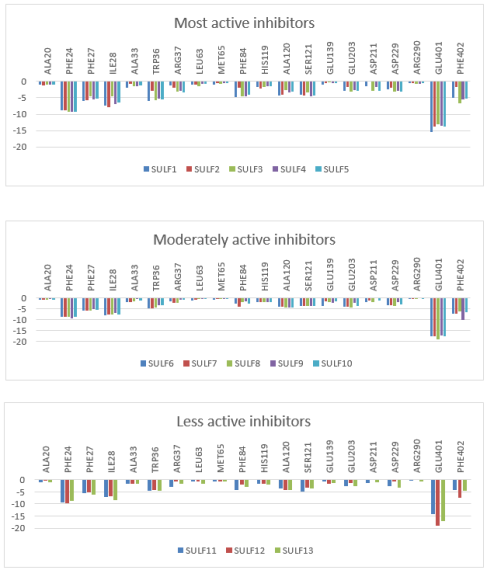
Figure 4. Per-residue decomposition of the intermolecular interaction energies (Eint) derived from molecular mechanics computations (values in kcal·mol-1). The charts group the derivatives by potency: (top) highly active compounds; ( center) intermediate activity analogs; and (bottom) low activity variants (Table 2 [4]).
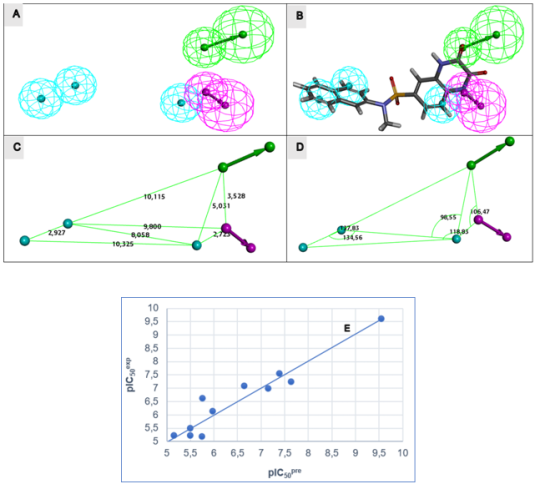
Figure 5. (A) Spatial arrangement of the generated features. (B) Superimposition of the optimal pharmacophore onto SULF1, the highest-affinity Eis inhibitor. (C) Distances between centers, (D) angles between centers of pharmacophoric features. Feature legend: HYD = Hydrophobic (cyan), HBA = Hydrogen bond Acceptor (green), HBD = Hydrogen bond Donor (pink), Excluded volume. (E) Correlation plot of experimental vs. predicted inhibitory activity.
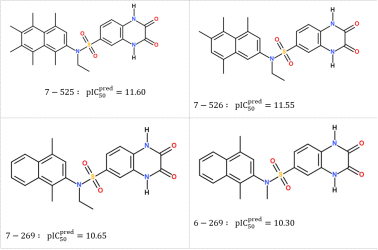
Figure 6. Chemical structures and predicted activities () of the most promising in silico designed SULF analogs.
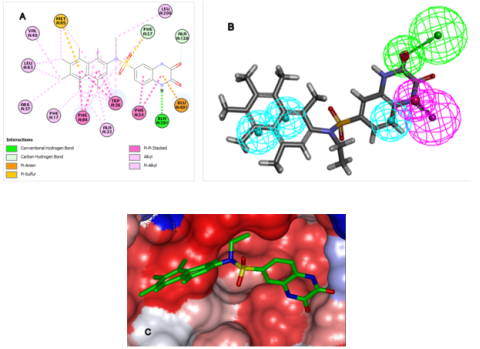
Figure 7. (A) Two-dimensional interaction map illustrating the optimal designed candidate, SULF 7-525, docked within the Eis catalytic pocket. (B) Spatial alignment of the SULF 7-525 derivative against the established PH4 inhibitory model. (C) Three-dimensional representation of the Eis active site’s Connolly surface accommodating the reference SULF 7-525 structure. (Color scheme: hydrophobic regions in red, hydrophilic areas in blue, and intermediate zones in white).
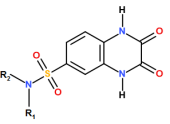
Figure 8. Core chemical scaffold of the sulfonamide (SULF) library illustrating the R1 and R2 substitution positions.
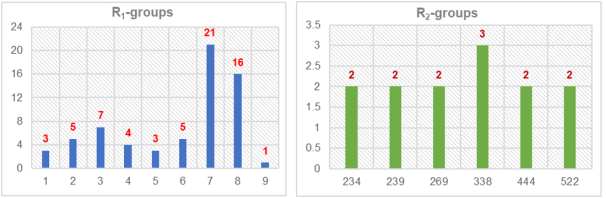
Figure 9. Distribution histograms illustrating the occurrence rates of specific R-substituents among the top 65 analog hits that successfully align with four essential components of the Hypo1 pharmacophore model.
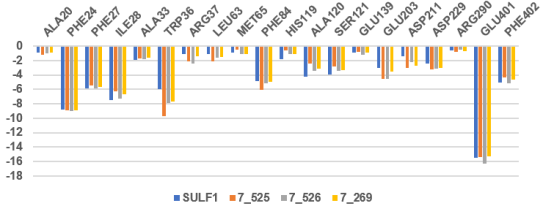
Figure 10. Per-residue partitioning of the computed intermolecular interaction energies (Eint in kcal.mol-1) comparing the three most favorable novel derivatives against SULF1, the reference high-affinity molecule from the TS. The color coding corresponds to the respective ligands.
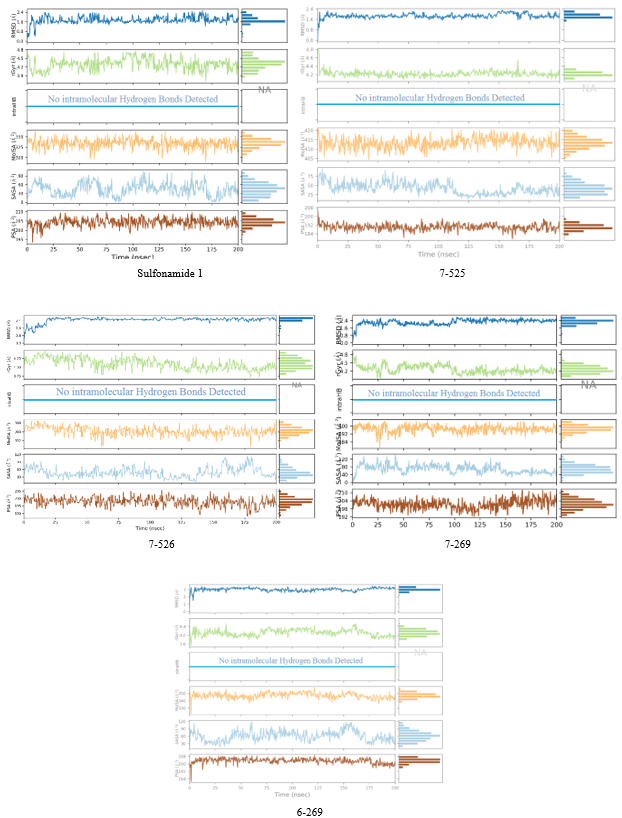
Figure 11. Trajectory analysis depicting the temporal variation of SULF-Eis complex characteristics throughout 200 ns MD simulations. For each evaluated ligand, the graphs display the following parameters (from top to bottom) measured against the simulation timeframe: conformational root-mean-square deviation (RMSD) relative to the reference state, gyration radius (rGyr), internal hydrogen bond occurrences (intraHB), as well as the molecular (molSA), solvent-accessible (SASA), and polar (PSA) surface areas. Sulfonamide 1(SULF 1): the most active ligand among the molecules used in the training set.
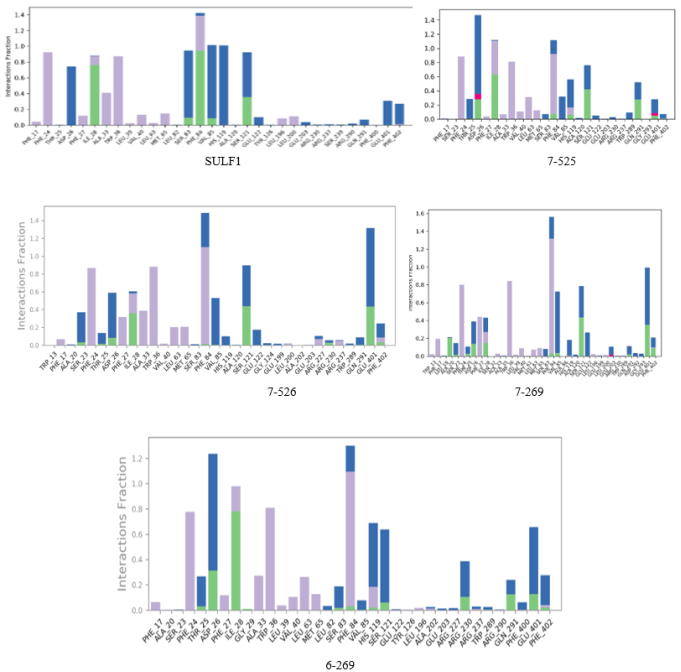
Figure 12. Partitioning of specific catalytic pocket residue interactions involved in anchoring the inhibitors within the SULF-Eis systems over the course of MD trajectories. Interaction types are color-coded as follows: hydrogen bonds (green), ionic bonds (magenta), hydrophobic associations (purple), and water-mediated bridges (blue).
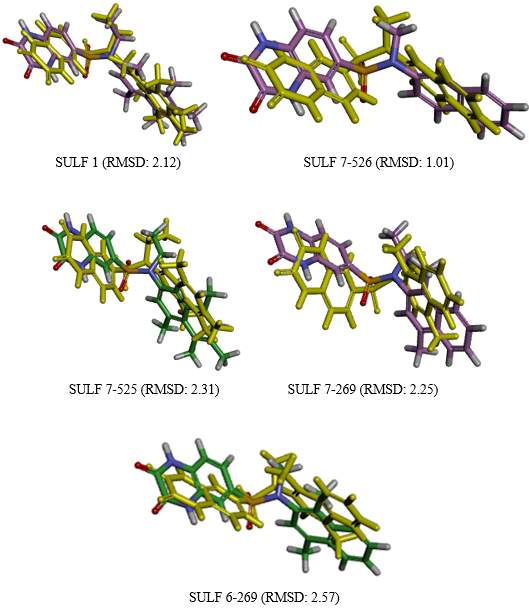
Figure 13. Superimposition of the functionally active ligand geometries optimized via molecular mechanics against the mean operational conformations (depicted in gold) extracted from the molecular dynamics trajectories.
Information


















