1. Introduction
Carbon Capture and Storage (CCS) is an established technology for carbon abatement. Moreover, in absence of better alternatives it presents a way forward there is sufficient sequestration capacity world-wide to accommodate emissions for over a century
. Currently, approximately 37 million tons per year (Mt/y) of CO
2 worldwide or about 0.1% of our global emissions (37 Bt/y) are effectively sequestered through CCS
. The main issue with CCS is its cost, which makes larger scale application of the technology challenging.
1.1. Notional Cost of Carbon-Neutral Energy
In establishing a benchmark for the cost of carbon-free energy, we consider the expense of energy derived from fossil fuels and rendered carbon neutral by employing CCS. Recognizing the cost of CCS varies greatly with geography
| [2] | Advisory Council, E. f. (2011). The Costs of CO2 Capture, Transport and Storage. European Technology Platform for Zero Emission Fossil Fuel Power Plants. |
| [3] | Alexander Zoelle, D. K. (2015). Cost and Performance Baseline for Fossil Energy Plants. USDOE NETL. |
| [4] | Anchondo, C. (2021, March 29). DOE lab unveils technology to slash CCS costs. Energy and Environment. |
| [23] | Maura Vaccarelli, R. C. (2014). Energy and economic analysis of the CO2 capture from flue gas of combined cycle power plants. Energy Procedia (pp. 1165-1174). Elsevier. |
| [20] | Irlam, L. (2017). GLOBAL COSTS OF CARBON CAPTURE AND STORAGE. Global CCS Institute. |
[2-4, 23, 20]
and taking a representative number of $C 138.5/tCO
2(net) and assuming a long-term average NG price of $C 2.5/GJ, carbon neutral heat at (e.g. gas fired power plants) can be obtained at $C10.1/GJ. This doesn't equate to the cost of carbon-neutral energy for retail use, however for point sources of emissions any new technology must compete with this yardstick for successful adoption.
1.2. CCUS and Issues
When CCS is accompanied with CO
2 utilization as in CO
2-EOR (or when CO
2 capture is necessary for natural gas purification), it is found to be economically viable. CCS with CO
2-EOR is a specific case of CCUS – Carbon Capture, Utilization, and Storage, and only about 0.5% of global oil production is amenable to CO
2-EOR
. If application of this form of CCUS is extended to all potential known suitable reservoirs (containing about 470Bbbl reserves
| [26] | Michael Godec, V. K. (2011). CO2 Storage in Depleted Oil Fields: The Worldwide Potential for Carbon Dioxide Enhanced Oil Recovery. Energy Procedia, 2162–2169. |
[26],
of the total remaining reserves of 1600Bbbl
) within reach of CO
2 production sites, its extent could cover about 7% of global emissions by 2040 (IEA SDS report), increasing from its current 0.1%. This is significant but still less than 10% of the challenge.
In its broader sense, CCUS involves capturing CO
2 from waste streams, and utilizing it products that would otherwise incur a cost, such as plastics, cement, food, activated carbon, soap, and fuel, etc. Can the remaining 90% emissions challenge be addressed with the general form of CCUS where useful products from it are expected to offset the high cost of underlying energy intensive processes? It’s easy to see if we convert even a fraction of CO
2 to useful products such as cement-substitute, the supply of such products will overwhelm the market, resulting in the loss of much of its market value, and making it impossible to recover costs. With one exception, we do not consume anything globally in that amount, as
Table 1 shows. In fact, top 10 commodities the world consumes on annual basis add up to less than 10 Bt. This implies when conversion to useful products is talked about in CCUS context, either the extent of application is assumed to be insignificant, or assessment of product value is unreasonably optimistic. In either case the CCUS approach of making useful products from CO
2 has limitations of application at a significant scale.
Table 1. Annual global production and consumption of various substances.
Products/Substance | Approximate amount, B t/y |
CO2 emitted | 37 |
Carbon in emitted CO2 | 9.8 |
Cement use | 4 |
Food consumption | 3.6 |
Steel use | 1.8 |
Plastics (PE, PP, PVC, etc.) use | 0.35 |
Asphalt consumption | 0.12 |
Natural rubber use | 0.014 |
Activated carbon use | 0.006 |
We consume about 12 B t/y of fossil fuel including oil, gas, and coal, overcoming the argument presented above. Arguably then, if all the emissions are converted back to fuel (using solar energy, and assuming technoeconomic feasibility), one can fix all the emissions. Additionally, by burning that fuel and carrying the process in a cyclical manner, carbon addition to atmosphere can be avoided. While it is technically feasible to do this, the cost of such fuel is always going to be more than the carbon-neutralized fossil fuel, as explained in Appendix I. This appendix also explains why converting CO2 using solar energy to fuel is also less energy efficient for transport purposes than using that solar energy directly to charge batteries to run, for example, the EVs. In summary, even the renewable-fuel route of CCUS is not economically robust.
Against this background the highlights of this paper include:
1. Description of the economic issue with CCS and the general form of CCUS, including the case of renewable fuels, as explained above
2. Identification and dealing with the underlying issue of using carbonaceous fuels (gaseous nature of combustion byproduct - CO2) directly
3. Presentation of novel lower-energy-investment approaches where the degree of carbon oxidation (oxidation number) can be controlled to yield only non-gaseous byproduct
4. Describing L-ox and L-red approaches which help achieve this by thermo-catalytic or electrochemical means, and providing literature references in support of the proposed chemical reactions
5. Identification of specific gaps for further development, to exploit these approaches
3. The Potential of Electro-Thermo-Chemical Methods in Carbon Abatement
There are two approaches discussed here that promise to be more cost effective than the alternatives on account of lower energy requirements in bypassing formation of gaseous byproduct of combustion – CO2 or converting it back to non-gaseous products, as well as requiring fewer unit operations and simpler equipment to carry out the process.
These two approaches broadly fall into the categories of (
1) avoiding formation of CO
2 in combustion process by keeping the end waste product in liquid / solid state through lower oxidation or L-ox, and (
2) if the combustion is carried to the fullest extent, converting the CO
2 back into a solid or liquid matter through lower reduction or L-red, with least energy input. The ensuing discussion answers if the two approaches are chemically feasible, i.e., if the proposed reaction routes resulting in non-gaseous byproducts of oxidation of fuels are feasible and proven.
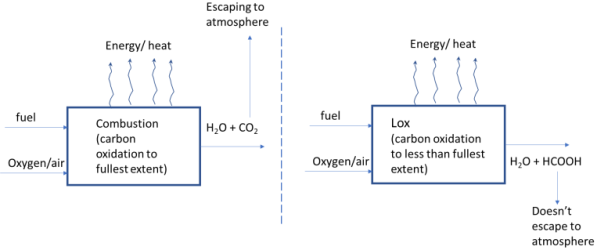
3.1. L-ox Processes
The concept of L-ox is also illustrated schematically in
Figure 1. In conventional combustion, fuel is combusted to the highest oxidation state of carbon. This results in formation of CO
2 which escapes to atmosphere. In Lower-oxidation (L-ox) the fuel is oxidized only to state of oxidation yielding a liquid or solid waste byproduct. Such waste products do not interact with the atmosphere and can be more easily disposed of.
Figure 1. Conventional combustion vs. L-ox.
While a hydrocarbon fuel can be represented chemically as CxHy, the simplest of these is CH
4 or methane. On combustion with oxygen, they produce water and CO
2 and yield the desired energy. This can be described by a stoichiometric reaction (
1) below:
(1)
For methane this simplifies to reaction (
1b)
For keeping the discussion simple only fuel is considered and it is assumed the same applies to other fuels with some modification.
3.1.1. Notional Economics
While a detailed economics of the process is beyond the scope of this work, a quick estimate of the benefit of the presented approach can be had with the following analysis. An observation from
Table 2 is that if the fuel is oxidized only to such degree that carbon attains an oxidation number of 2, one will be able to keep the product of oxidation (HCOOH) in liquid state and be able to still withdraw approximately 70% of the energy contained in CH
4 and leave a waste byproduct that does not result in emissions. This is shown in the reaction (
2).
(2)
HCOOH is an example of lower oxidation, but any other non-gaseous product having lower oxidation of carbon than CO2 such as oxalic acid is going to be useful. The cost of carbon-neutral energy, before accounting for Opex and Capex of the process, becomes $C 3.66/GJ (compared to $C 2.5 with emissions). The challenge in presenting actual economics is in the estimation of Capex and Opex in absence of an established technology for this process at present. To overcome this predicament, one can make some reasonable assumptions for these two costs based on appropriate analogues. These two together are assumed here to be equivalent to an additional 50% cost of lost energy (per GJ). It can be argued that 50% is too little or too much for this contingency, but a reasonable place holder can be assigned for a preliminary assessment of the opportunity, and revised when more is known about the detailed process equipment.
The implicit cost of carbon abatement in leaving the end oxidation product as HCOOH becomes $21/tCO2 [= (3.66-2.5)/0.055]. With the 50% additional assumed, this becomes $32/GJ. This compared with the CCS cost of $115/tCO2-processed (or $138/tCO2-net) - a difference of $83+/tCO2 is thus the incentive for pursuing this route to carbon abatement. Carbon neutral heat with these assumptions is $4.3/GJ as opposed to $10.1 estimated above with CCS.
3.1.2. One-Step Conversion vs. Two-Step
Ideally, to replace the simple combustion reaction (
1b) by (
2), it will be preferable to have a process to do it in one step, such as the one described by Li et al.
| [25] | Mengwei Li, J. S.-S. (2021). Single-step selective oxidation of methane to methanol in the aqueous phase on iridium-based catalysts. Applied Catalysis B: Environmental, 292 (September). |
[25]
. It is surmised that the reason why more literature on it is not available, could be the difficulty to control the oxidation from proceeding to completion to make CO
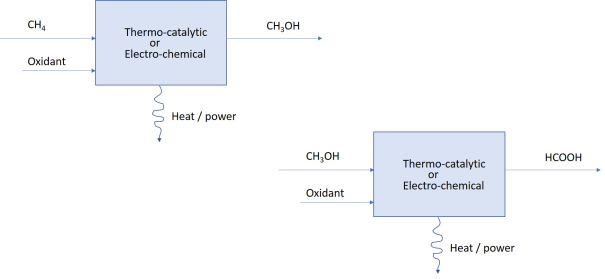
2. That is why a two-step oxidation process is considered here. In the first step methane is oxidized into methanol. This releases part of the available energy as heat or power. In the second step methanol is oxidized into formic acid, giving up the remaining extent of energy without leaving a gaseous waste product - CO
2. For the stated purposes these individual steps are not burdened with the requirements of producing pure methanol or other intermediate-oxidation products such as aldehydes and ketones. The objective is to extract energy, not the purity of the product. The two steps are shown in the reaction (
3).
(3)
Although from energy extraction perspective a one-step process (
2) may be more efficient, there are some commercial benefits of carrying out the process in two steps denoted by (
3). A significant number of natural gas reservoirs found the world are isolated and too small to be commercially exploited. For commercial exploitation one needs these pools to be either connected to a pipeline network which is expensive, or compressed as CNG or LNG and transported by road/rail, which is also expensive. If the produced gas could be converted to a liquid fuel economically such as CH
3OH, it will greatly enhance the commercial viability of these gas pools. CH
3OH is a commercial product in its own right, and the current commercial process to convert methane to methanol is highly capital and energy intensive. Secondly, methanol being a liquid fuel, it can find use relatively more easily in transport sector either through Direct Alcohol Fuel Cell (DAFC) or though modification of ICEs (MICE), requiring relatively a smaller degree of change in the existing infrastructure. There is a potential (at least thermodynamically) to exploit the thermal energy released in conversion of CH
4 and at the same time make a more valuable product, with least amounts of emissions in step – I, CH
4 to CH
3OH. Similarly, in step-II (CH
3OH to HCOOH), there is a potential with a modified version of DAFC (MDAFC) to exploit the energy without emitting CO
2. However, all currently discussed DAFCs in literature result in CO
2 emissions, just as current ICEs result in complete combustion producing CO
2. MDAFC or MICE will require innovation to prevent ultimate oxidation of the fuel to CO
2 and only result in liquid (waste) product – HCOOH. This concept is illustrated schematically in
Figure 2.
Figure 2. Energy without emissions: two-step process.
As indicated above, these two energy-releasing steps can either be carried out through a process based on thermo-catalysis, or on electro-chemistry. The following discussion is dedicated to substantiate the feasibility of step I and II processes with based on the existing literature, and pointing out the areas where further work is needed to complete the technological gaps.
3.1.3. Thermo-catalytic L-ox
(i). Step-I: CH4 to CH3OH
Zuo et al.
| [36] | Zhijun Zuo, P. J. (2016). The Low‐Temperature Conversion of Methane to Methanol on Ce‐Ox/Cu2O catalysts: Water Controlled Activation of the C‐H Bond. Brookhaven National Laboratory, USDOE. |
[36]
describe a low temperature reaction at 450K in presence of water, and CeO
2/Cu
2O/Cu(III) catalyst that converts CH
4 to CH
3OH with high selectivity. Li et al.
| [25] | Mengwei Li, J. S.-S. (2021). Single-step selective oxidation of methane to methanol in the aqueous phase on iridium-based catalysts. Applied Catalysis B: Environmental, 292 (September). |
[25]
discuss an iridium-based catalyst system (Ir-ZSM-5) in aqueous phase in presence of CO that converts CH
4 either to CH
3OH or to HCOOH depending on the addition of a second metal. With Cu the system selectively produces CH
3OH, and with Pd formic acid (HCOOH) is produced. They report methanol selectivity to be in the range of 80% at 150 C in a IrCuPd-ZSM-5 trimetallic catalyst. The one step conversion from CH
4 to HCOOH mentioned by Li et al. is right along the most favored path stated above in reaction (
2).
Although thermodynamically favored exothermicity of these reactions, larger scale experiments need to be carried out to establish availability of usable high-grade heat. This is also true of the step-II described below.
(ii). Step-II: CH3OH to HCOOH
Many online available texts on chemistry, e.g.,
, or
give a catalytic route to conversion of methanol to formic acid in presence of sodium dichromate (Na
2Cr
2O
7) and sulfuric acid (H
2SO
4). However, from these sources do not specify how that heat manifests itself. The two-step reaction is as given by (
4) and net reaction as shown in (
5).
(5)
While in presence of these catalysts the reaction can achieve the desired conversion of methanol to formic acid, methanol can also be oxidized in presence of silver (Ag) catalyst at high temperature (500C) with air
. This converts methanol to methanal (HCHO). This is in fact the commercial process for making HCHO from methanol. However not enough in the literature is available to suggest that the process can be extended to further oxidation of HCHO to formic acid releasing full extent of intended heat. This is an area for further investigation. A thermo-catalytic process that converts methanol to HCOOH will look similar in terms of the equipment to the combustion process. The combustion process typically used in fired heaters (or boilers) has air and fuel reacting at the specially designed burners. The outside surface of the tubes provides the heat transfer area. In the L-ox case, burner may be replaced with a mixer, but the heat transfer area will be designed to also to provide the catalytic action needed for the reaction to limit the oxidation to just HCOOH.
3.1.4. Electrochemical L-ox
The process of electrochemical conversion of CH4 to HCOOH is thermodynamically favored to generate electric power in a galvanic cell directly, as is accomplished in fuel cells, bypassing the step of first generating the heat and converting it to power with heat engines. The efficiency of the latter is dependent on the grade (temperature) of the generated heat as dictated by Carnot’s law. The mechanics of the cell operation may additionally provide reaction controls to limit oxidation of the fuel to only a certain oxidation number. Again, the feasibility of conversion of CH4 to HCOOH and generating useful energy is not completely established but available literature points to the potential of the process with some further development.
(i). Step-I: CH4 to CH3OH
Haomin
| [17] | Haomin Jiang, L. Z. (2022). Direct conversion of methane to methanol by electrochemical methods. Green Energy & Environment, 7, 1132-1142. |
[17]
, presents a good summary of advances in electro-catalytic conversion of CH
4 to CH
3OH and the challenges in this process. Yanfang Song
| [34] | Yanfang Song, Y. Z. (2020). Efficient methane electrocatalytic conversion over a Ni-based hollow fiber electrode. Chinese Journal of Catalysis, 41(7), 1067-1072. |
[34]
and Zhikai Guo
| [37] | Zhikai Guo, W. C. (2020). Electrocatalytic oxidation of methane to ethanol via NiO/Ni interface. Applied Catalysis B: Environmental, 270(August). |
[37]
mention conversion of CH
4 to ethanol with 89% Faradaic efficiency (FE) with NiO/Ni catalyst at ambient temperature and pressure and alkaline electrolyte. While this is a great discovery from the perspective of monetization of isolated gas pools as well as to meaningfully use the otherwise flared associated gas, it is unfavorable from carbon abatement perspective - where the aim is to exploit maximum energy out of the fuel and leaving minimal amount with the waste byproduct. Ethanol does not qualify for doing that. But if ethanol could again be used in the second step as feed to yield HCOOH (or e.g., oxalic acid etc.) and energy, it will be a desirable scenario. With slightly different conditions they also report conversion to CH
3OH with 54% FE. Kim et al.
| [30] | Soyoung Kim R., and Surendranath, Y. (2019). Electrochemical Reoxidation Enables Continuous Methane-to-Methanol Catalysis with Aqueous Pt Salts. ACS Central Science, 5, 1179-1186. |
[30]
discusses a platinum- based catalyst which at moderate temperature results in continuous CH
3OH production with 70% selectivity. Maria Sarno et al.
| [22] | Maria Sarno, E. N. (2020). Methane electrochemical oxidation at low temperature on Rh single atom/NiO/V2O5 nanocomposite. Applied Catalysis A: General, 603(August). |
[22]
used a nano catalyst (nanocomposite) based on NiO and V2O5 with Rh dispersed (NiO-V2O5/Rh) at 100C temperature, resulting into methanol formation with 91% FE and 97% selectivity.
An important issue to note here is that even if the electrochemical systems described here have shown higher selectivity to make the desired product, this is not achieved in a galvanic-cell operation. The reason is that the provision of active oxygen here generally comes from splitting water (or another reactant giving active oxygen) at the cathode. This means the reaction at the other electrode will be even more spontaneous with giving more energy. However, to establish that the overall cell results in power generation (rather than some power input and a lot of heat output) still remains to be demonstrated.
(ii). Step-II: CH3OH to HCOOH
In the context of ethanol, many scientists such as Ye Wang
| [35] | Ye Wang, S. Z.-B. (2015). Recent Advances on Electro-Oxidation of Ethanol on Pt- and Pd-Based Catalysts: From Reaction Mechanisms to Catalytic Materials. Catalysts, 5, 1507-1534. |
[35]
or El Mahdi Halim
| [8] | El Mahdi Halim, S. C. (2022). Recent Advances in Anode Metallic Catalysts Supported on Conducting Polymer-Based Materials for Direct Alcohol Fuel Cells. Frontiers in Energy Research, 10(March). |
[8]
have described its electro oxidation with Pt or Pd Catalysts follows a dual pathway. Under C1 path results in CO
2 but under C2 path results strictly in CH
3COOH. Formation of CH
3COOH appears to be a dead-end reaction, with acetic acid not reacting further. This encourages a surmise that methanol to HCOOH formation with similar arrangement may be possible, if there were an equivalent C2 path in this system.
Xinfa et al.
| [33] | Xinfa Wei, Y. L. (2020). Formic Acid Electro-Synthesis by Concurrent Cathodic CO2 Reduction and Anodic CH3OH Oxidation. Angewandte Chemie Internationa Edition, 60(6), 3148-3155. |
[33]
, amongst many other authors, describe formation of HCOOH from CH
3OH at anode, however at cathode the usable (active) oxygen comes from reduction of CO
2. This is a similar situation to the one described under the step-I, as for the needed reduction of carbon at cathode to yield [O] electric power needs to be fed into the circuit. Ideal system would be able to use oxygen from air, avoiding needed energy for the CO
2 reduction reaction.
With the current search, there are not too many indications of a lab result about electro-oxidation of CH3OH to HCOOH in the literature. In absence of missing electrolytic step-II, the possibility still exists to exploit the step-I potential electrocatalytically and carry out the second step with thermo-catalytical methods.
To recap the gaps in the electrochemical route:
1. Ideally there should be a single step conversion of CH4 to HCOOH yielding energy enabled by use of oxygen from air
2. Step-I while feasible needs to be improved to be able to use oxygen from air
3. Step-II needs discovery of suitable catalysts and electrolyte system for making it feasible
3.2. L-red Processes (Low Energy Reduction of Combustion Byproduct - CO2)
3.2.1. Preservation of Produced Biomass as a Means of Carbon Sequestration and Challenges
A subclass of L-red process deserves some consideration where CO
2 is converted to biomass naturally using solar energy. The energy required in making the renewable fuels from CO
2 is exactly as much as we took out by burning the fuel, plus some additional amount to carry the process out. This is obviously highly energy intensive as discussed above. But according to
Table 2, we have number of other options. If we convert CO
2 back to complex sugars (carbohydrates) mentioned in step 2, we need to supply only one third energy compared to converting it all the way back to being a fuel, and at the same time we have the carbon sequestered in solid form! This is essentially the idea behind enhanced biomass growth as a means of carbon sequestration, or afforestation, or bio-sequestration. Again, the energy needed for converting CO
2 to biomass comes from the sun, and the process occurs naturally all around the globe where temperatures are between 0 and 40
o Celsius, and water is not scarce. The issue with this approach is that produced biomass is subject to insect and fungal activity as a result of which carbon stored in biomass is eventually release back to the atmosphere as CO
2. But with preservation of biomass, this hurdle can be overcome. Appendix II describes several approaches for preservation of biomass for long term carbon sequestration.
3.2.2. Exploring Feasibility of Electro-chemical L-red
One limitation with biomass-preservation as a sequestration-approach is its dependence on procuring or growing sufficient biomass which can be expensive due to the process being area intensive and slow, even if the technology exists.
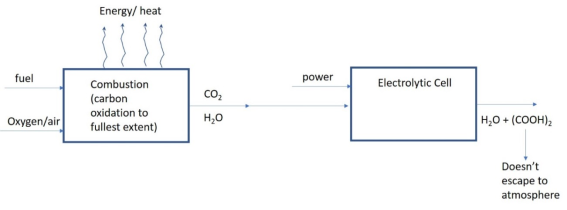
If the produced combustion gas after cooling (CO
2 + N
2) is routed in an electrochemical cell along with water where CO
2 preferentially passes through the semi-permeable barrier, and N
2 acting as an inert, and the other inputs such as electrolyte solution and catalysts are chosen properly, CO
2 can be converted (reduced) to, for example, oxalic acid [(COOH)
2] with relatively small amount of energy. This is illustrated in
Figure 3.
Figure 3. L-red process concept explained.
Heat of combustion of oxalic acid is approximately 2.8 GJ/t or 252 GJ/t-mole, or 2.8GJ/tCO2, implying 2.86GJ energy needs to be supplied to reduce 1 t CO2 to oxalic acid. This energy however is going to be supplied in the form of electric power for the cell to function. The cell will have to operate in a manner that a constant stream of (COOH)2 will have to be withdrawn constantly as waste byproduct to maintain a certain concentration of reactants in the cell. The cost of such process will be on account of power input and capex + opex of the cell. If the energy efficiency of the cell is 80%, cost of power is $60/MWh, on account of power input alone, the cost of carbon abatement will be (2.86GJ/t)/(3.6GJ/MWh)x60/0.8 = $59.6/tCO2. The remaining cost can only be guessed at this juncture and assuming it to contribute to about half of the power cost over the usable life of the cell, the total cost of carbon abatement will be approximately $90/tCO2. Oxalic acid being unmonetizable (even with a few uses) due to its large scale for reasons described above, does not need to incur costs for further transport and sub-surface injection. It can be surface stored.
If one were to convert CO2 to other substances such as carbon fiber, or plastics, or complex sugars, the cost of input power will be relatively higher. For pure carbon (30GJ/t C) with 80% efficiency, it will be (12/44)*(30/0.8)*(60/3.6) + 30 = $201/tCO2. If CO2 were to be converted to biomass (17.3GJ/t) or complex sugars [(CH2O)x] electrochemically with 80% efficiency, the cost will be (30/44)*(17.3/0.8)*(60/3.6) + 30 = $276/tCO2. All said though, the potential of this alternative needs to be established further with additional research and development.
If L-red cell only accepts pure CO
2 then $115*0.75 ~$86/tCO
2 will be used for capture and ~$90/tCO
2 for its conversion to oxalic acid for a total of ~$176/tCO
2 sequestered. This obviously does not compare favorably with the $115/tCO
2 cost of CCS. This suggests the cell has to be such that flue gas mixture can enter the unit without needing CO
2 separation. In support of such reactions being technically feasible, Weixin et al.
| [32] | Weixin Lv, R. Z. (2013). Electrochemical reduction of carbon dioxide with lead cathode and zinc anode in dry acetonitrile solution. Solid State Electrochem, 17(July), 2789–2794. |
[32]
report a combination of lead cathode and a sacrificial Zn anode in 0.1TEAP/AN electrolyte leads to formation of insoluble (and more easily recovered) Zinc Oxalate with 88.7% efficiency at 2.2 to 2.8V. In their experiment CO
2 was bubbled into the cell. Interestingly when N
2 was bubbled there was no reduction wave in the voltammograms, pointing to a potential of introducing a mixture of CO
2 and N
2 without having to separate the two in advance.
Ito et al.
| [21] | Ito, K. I. (1985). Electrochemical Reduction Products of Carbon Dioxide at Some Metallic Electroded in Nonaqueous Electrolytes. Bull. Chem. Soc. Jpn., 58(October), 3027-2028. |
[21]
describe use of lead cathode at 3V with TEAP/PC non-aqueous electrolyte, oxalic acid was formed with 80% current efficiency, which decreased with increasing voltage. Ikeda et al.
| [18] | Ikeda, S. T. (1987). Selective Formation of Formic Acid, Oxalic Acid, and Carbon Monoxide by Electrochemical Reduction of Carbon Dioxide. Bull. Chem. Soc. Jpn., 60 (July), 2517-2522. |
[18]
describe use of various electrode and electrolyte combinations to reduce CO
2 to formic or oxalic acid with very high current efficiencies. With lead cathode (working also as a catalyst) in aqueous electrolyte TEAP/H
2O (0.1M tetraethylammonium perchlorate in water), at 2.4V potential formic acid was formed at 78.9% efficiency. With Indium cathode at 2V formic acid was formed with 87.6% efficiency. Similarly, with TEAP in non-aqueous solution comprising propylene carbonate (PC), acetonitrile (AN), and dimethyl sulfoxide (DMSO), the reaction favored formation of oxalic acid. With Pb cathode and 2.6V, oxalic acid formation was with 73.3% current efficiency.
While these results are extremely promising, they do not completely present a workable technology. Further work focused on the following areas is needed:
1. Introduction of N2/CO2 mixture in the cell to establish and obviate the need for CO2 separation
2. Establish impact of NOx/SOx on the conversion process and the equipment
3. Increase conversion efficiency further to 95+% by using specially designed catalysts
As indicated above the energy needed to convert CO2 to oxalates is going to be the least and that is why it is best to not to attempt making useful products out of CO2. ‘Useless’ in this case is of a lot of use as it requires significantly less energy.